Bei gegebenem Druck ist immer die Phase in einem bestimmten Temperaturbereich stabil, deren chemisches Potential dort niedriger als das jeder anderen Phase ist. Die niedrigsten chemischen Potentiale findet man für Festkörper; und damit ist die feste Phase bei tiefen Temperaturen die stabilste. Da jedoch die chemischen Potentiale der einzelnen Phasen in unterschiedlicher Weise von der Temperatur abhängen, kann bei Temperaturerhöhung das chemische Potential einer anderen Phase (einer weiteren festen, einer flüssigen oder auch einer gasförmigen) unter den Wert für den Festkörper fallen. In diesem Fall findet ein Phasenübergang statt (u. Umständen extrem langsam, wenn eine kinetische Hemmung vorliegt).
Temperaturabhängigkeit
der Stabilität von Phasen
Die Temperaturabhängigkeit der Freien Enthalpie eines Systems
als Funktion der Entropie ist: (∂G/∂T)p
= −S. Da das chemische Potential eines reinen
Stoffes seiner molaren Freien Enthalpie entspricht (µ = Gm
= G/n), kann man die Temperaturabhängigkeit
des chemischen Potentials als Funktion der molaren Entropie schreiben:
| (∂µ/∂T)p = −Sm. |
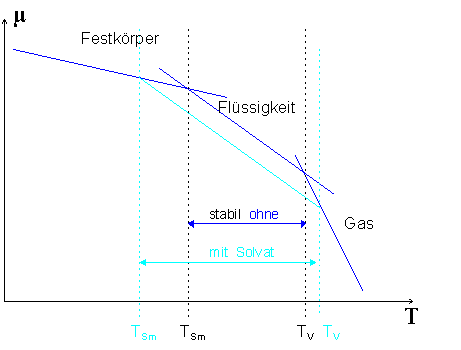 |
| Abb.1:
Chemisches Potential µ für drei Phasen. Die Steigung ist durch
-Sm gegeben. Mit Solvat
verschiebt sich µ(l) zu niedrigeren Werten mit entsprechender
Änderung des Schmelzpunktes TSm
und des Siedepunktes TV.
(Eigentlich müssten die einzelnen Geraden leicht nach unten gekrümmt sein, da die Entropie mit der Temperatur zunimmt. Zur klareren Darstellung ist aber darauf verzichtet worden.) |
Die Phasengrenzlinien
Zwei Phasen, die sich miteinander im Gleichgewicht befinden, besitzen
das gleiche chemische Potential. Für zwei Phasen α
und β bedeutet das
| µα(p, T) = µβ(p, T); |
die Lösung dieser Beziehung ist die Gleichung der Phasengrenzlinie in Form einer Funktion p(T). Ein charakteristisches Merkmal der Phasengrenzlinien ist deren Steigung dp/dT. T und p sollen sich in infinitesimalen Schritten so ändern, dass das Gleichgewicht zwischen α und β stets aufrechterhalten bleibt. Zu Beginn sind die chemischen Potentiale beider Phasen gleich (da das Gleichgewicht eingestellt ist). Wenn man sich auf die beschriebene Weise zu einem andern Punkt der Phasengrenzlinie bewegt, ist diese Beziehung immer erfüllt (da das Gleichgewicht eingestellt bleibt). Daher kann man die Änderung des chemischen Potentials beider Phasen gleichsetzen, dµα = dµβ. Wir wissen, dass für jede der beiden Phasen gilt dG = - SdT + Vdp. Division durch n ergibt:
Hier sind Smα und Smβ die molaren Entropien und Vmα bzw. Vmβ die molaren Volumina der jeweiligen Phase. Durch Zusammenfassen und Umformen dieser Gleichung erhalten wir
und daraus direkt die Clapeyronsche Gleichung
| dp/dT = ΔSm/ΔVm. |
Darin sind ΔSm = Smβ−
Smα und ΔVm
= Vmβ− Vmα
die Änderungen der molaren Entropie und des molaren Volumens des Systems
während des Phasenübergangs. Diese wichtige Beziehung für
die Steigung der Phasengrenzlinie gilt exakt und für jedes beliebige
Phasengleichgewicht eines beliebigen reinen Stoffes.
Die Phasengrenzlinie fest/flüssig
Der Schmelzvorgang findet bei einer Temperatur T statt und verläuft
unter Änderung der molaren Enthalpie des Systems um ΔSmH.
Daraus ergibt sich die molare Schmelzentropie zu ΔSmH
/ T; die Clapeyronsche Gleichung für diesen Vorgang lautet
| dp/dT = ΔSmH/TΔSmV |
ΔSmV gibt die Änderung des Molvolumens beim Schmelzen an. Die Schmelzenthalpie ist (mit Ausnahme von Helium-3) immer positiv, die Volumenänderung ist normalerweise ebenfalls positiv und stets sehr klein. Dies bedeutet, dass die Steigung dp/dT steil und im allgemeinen positiv wird. Die Funktion p(T) selbst erhält man durch Integration von dp/dT unter der Annahme, dass ΔSmH und ΔSmV nur wenig von der Temperatur abhängen und daher als Konstanten behandelt werden können. Wenn die Schmelztemperatur bei dem Druck p* gleich T* und bei dem Druck p gleich T ist, haben wir folgende Integration durchzuführen:
p*∫p dp = ΔSmH/ΔSmVT*∫T (dT/T).
Wir (James Thomson war der Erste) erhalten als Näherungsgleichung
für die Phasengrenzlinie fest/flüssig
| p = p* + ΔSmH/ΔSmV ln (T/T*). |
Für kleine Temperaturdifferenzen T - T* erhalten wir ln(T/T*)
= ln(1 + (T - T*)/T*)
≈(T
- T*)/T*
(wegen ln(1+x) ≈ x für kleines x):
| p ≈ p* + ΔSmH/ΔSmV (T - T*)/T*. |
Die Phasengrenzlinie flüssig/gasförmig
Die molare Verdampfungsentropie eines Stoffes bei der Temperarur T
beträgt
ΔVH/T; die Clapeyronsche
Gleichung der Phasengrenzlinie demzufolge
dp/dT = ΔvH/TΔvV.
Die Verdampfungsenthalpie ist stets positiv, ΔVV ist ein großer, ebenfalls positiver Wert. Die Ableitung dp/dT ist daher auch positiv; aber wesentlich kleiner als für die Phasengrenze fest/flüssig.
Das Molvolumen eines Gases ist viel größer als das einer
Flüssigkeit, so dass wir in guter Näherung ΔVV
≈
Vm(g) setzen können. Setzt man diese beiden Näherungen
in die exakt gültige Clapeyronsche Gleichung ein und nimmt ferner
ideales Gasverhalten (Vm(g) = RT/p) an, d.h. dp/dT
= ΔH p/RT² ,
dann erhalten wir die näherungsweise gültige Clausius-Clapeyronsche-Gleichung:
| d ln p/dT = ΔVH/RT2 |
(Dazu haben wir dp/p = d ln p verwendet.) Wenn wir noch voraussetzen,
dass die Verdampfungsenthalpie nicht von der Temperatur abhängt,
können wir diese Gleichung integrieren und erhalten
| p = p* exp[ΔVH/R(1/T* - 1/T)] |
Hier ist p* der Dampfdruck bei der Temperatur T*, p gehört entsprechend
zu T. Diese Funktion beschreibt den Verlauf der Phasengrenzlinie flüssig/gasförmig;
sie endet bei der kritischen Temperatur Tkrit, da oberhalb dieser
keine flüssige Phase mehr existiert.
Die Phasengrenzlinie fest/gasförmig
In allen für die Phasengrenze flüssig/gasförmig abgeleiteten
Beziehungen tauschen wir einfach die Verdampfungsenthalpie gegen die Sublimationsenthalpie
ΔSubH
aus. So erhalten wir, analog zur Herleitung der Clausius-Clapeyronschen
Gleichung, die folgende Beziehung für die Temperaturabhängigkeit
des Sublimationsdampfdrucks:
| d ln p/dT ≈ ΔSubH/RT2. |
| p = p* exp[ΔSubH/R(1/T* - 1/T)] |
Da die Sublimationsenthalpie ΔSubH größer als die Verdampfungsenthalpie ΔVH ist, kann man dieser Gleichung entnehmen, dass in der Nähe des Schnittpunktes beider Kurven die Sublimationsdruckkurve steiler ansteigt als die Dampfdruckkurve.
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.