|
|
These processes appear to be different but in fact they are all closely
related. We would like to examine these in much greater depth to understand
them and their relationships.
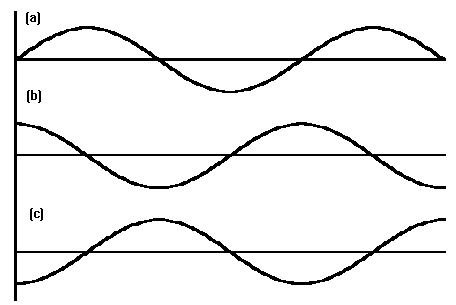
Curve (a) represents the oscillatory behavior of the spring, curve (b) is the applied force that is required to make it oscillate and curve (c) is the applied force that is required to stop it from oscillating. In (b) the spring is absorbing energy and in (c) it is providing energy. (b) represents absorption and (c) represents stimulated emission. The only difference is the phase - 180 degrees to be exact. Both (b) and (c) are out of phase with (a).
Our description so far has applied only to a driving force in resonance
with the vibrational frequency of the spring. What happens out of resonance?
If the driving frequency is lower than resonance, there will be an in-phase
component of the oscillation. If it is higher, there will be a 180 degree
out-of-phase component. If the driving force is a cosine function, the
oscillation will have a sine functionality at resonance and an additional
cosine functionality at higher or lower frequencies.
A series of contour plots representing the temporal behavior:
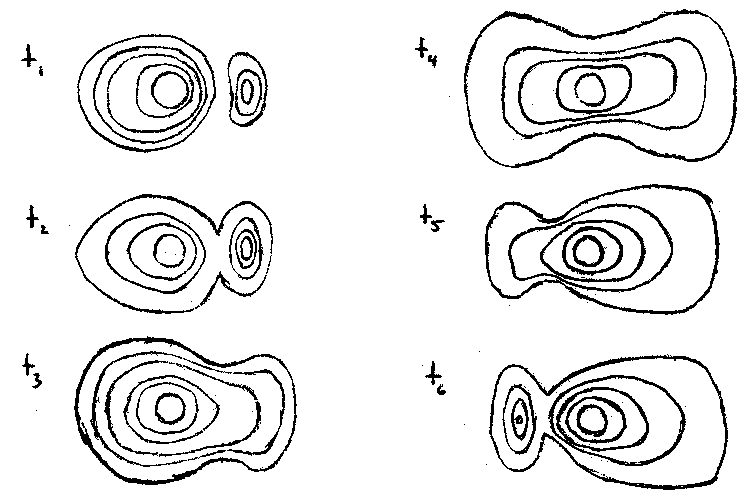
We start at t1 with a state that is primarily 1s and progress to a state at t4 that is primarily 2p and then back again. Quantitatively, we could use the time-dependent wave equation and derive a correct time dependent wavefunction for a two-state system that is interacting with an incoming electromagnetic wave whose frequency was w.
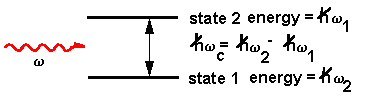
The Hamiltonian representing the interaction of the electric field, E, of the incoming wave with the dipole moment of the atom, µ, is:
H = µ E cos w t
The wavefunction as a function of time is then: (sorry too complicated
for HTML)
As an electromagnetic field propagates through a sample, there will be a certain average excited state population that will have a coherent and an incoherent component. At all times, the excited state has the option of simply relaxing spontaneously and emitting a photon. This process is spontaneous emission and is incoherent relative to the driven polarization. If the spontaneous emissions occurs from the coherent excited state population, the emission is called Raman scattering. The state it relaxes to can be a different electronic state in which case it is electronic Raman scattering, a different vibrational state where it is vibrational Raman scattering (or just Raman scattering), or a different rotational state where it is rotational Raman scattering. The emitted light frequency will differ from the exciting frequency by the energy of the final state. Quantum mechanically, Raman scattering is diagrammed by:
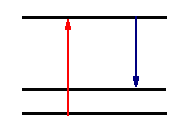
The top dotted line represents the coherent oscillating state. We know what that looks like. It is simply the atom or molecule distorted by the electric field of the incoming light wave. In a real system, it doesn't usually correspond to a particular stationary state of the molecule and since one doesn't usually want to have to derive the eigenfunctions that describe the state, we instead simply express it as a linear combination of all the states required to make this distorted state. It is called a virtual state because there is no real state at that energy usually. But we know it is simply the distorted molecule. Notice that the importance of Raman scattering depends upon how easily the molecule is distorted. Classically, this property is measured by the polarizability of the molecule and therefore polarizability determines the amount of Raman scattering.
Things become very interesting when the incoming electromagnetic wave approaches an electronic resonance. There is now the possibility of achieving a finite incoherent excited state population as well as a coherent population. The distortions of the molecules will become much larger near electronic resonances. Spontaneous emission will still occur from both the coherent and incoherent excited state populations which will now be considerably larger than when the driving frequency was far from resonance. The emission from the coherent population will be resonantly enhanced Raman scattering (resonance Raman) while the incoherent population will cause normal fluorescence. There will also be losses from the excited state population due to nonradiative relaxation. All of these losses result in absorption of light.
The relative amounts of resonance Raman and fluorescence depends upon the relative coherent and incoherent excited-state populations. This in turn depends upon the relaxation effects that are important in the system. In most molecular systems, dephasing by collisions or other interactions is a dominant factor in determining the incoherent population. The ratio of fluorescence to Raman will be approximately 2 / (T2 · γ) where T2 is the dephasing time constant and γ is the radiative rate of spontaneous emission.
The fluorescence that one observes can also have different character. If the fluorescence originates from an incoherent population that has been formed by dephasing, emission to the ground state will be resonance fluorescence and will have a lifetime characteristic of the emitting state. The linewidth of the emission will reflect the widths of the initial and final states. In contrast, the resonance Raman emission will cease when the excitation source is turned off and will have an emission linewidth characteristic of the final state. It is also possible that the incoherent excited population can be formed from relaxation from the state that was connected to the ground state. This fluorescence is relaxed fluorescence and is generally shifted in wavelength.
Either fluorescence or resonance Raman can be quenched. If there are
rapid nonradiative relaxation processes that depopulate the incoherent
excited state population, the fluorescence will be quenched. One would
then see only the resonance Raman. (The resonance Raman intensity could
also be lowered.) If there were rapid dephasing or relaxation processes
that depopulated the coherent populations, the resonance Raman emission
would be quenched. For example, one can observe strong Raman scattering
many times when the v = 0 to v = 0 electronic transition is excited but
very little when the v = 0 to v = n transitions are excited because the
vibrational states relax very quickly to the unexcited vibrational state.
Then the excitation profile of the resonance Raman effect will not follow
the absorption profile for the molecule.
Adapted from lecture notes by Prof. John Wright, Univ. of Wisconsin.
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.