Es existiert immer noch so etwas wie eine "Technologie-Lücke" zwischen den sehr detailierten Experimenten, die wir gerne durchführen würden, und denen, die wir durchführen können. In diesem Abschnitt widmen wir uns zunächst den wichtigsten Elementen eines idealen Experiments und schauen dann, wie weit die heute verfügbaren Methoden diesem Ideal nahekommen.
Wünsche
Um die Diskussion zu vereinfachen, beschränken wir unsere Aufmerksamkeit
auf eine Elementarreaktion, die nur drei Atome unfaßt, sagen wir
H + ICl → HI + Cl
Selbst in diesem einfachen Fall gibt es dazu eine energetisch mögliche Alternative, nämlich
H + ClI → HCl + I
Außerdem kommen gleichzeitig nicht-reaktive, elastische und inelastische Stöße vor. Für jeden der Reaktionswege kann das Halogenatom in seinem Grundzustand (2P3/2) oder im angeregten Feinstrukturzustand (2P1/2) gebildet werden; das entstehende Diatom wir zwar im elektronischen Grundzustand (X1Σ) sein, kann aber in jedem der vielen energetisch erlaubten Schwingungs-Rotationszustände vorliegen.
Eine vollständige Festlegung des Stoßvorgangs verlangt also nicht nur die (chemische) Identifizierung des Produkts (HCl oder HI), sondern auch die Feststellung des inneren Zustands von Edukten und Produkten, d.h.
H(2S) + ICl(v, j) → HI(v',j') + Cl(2P1/2)
und dazu die genaue Kenntnis der Stossenergie. In einem "noch idealeren" Experiment mit orientierten Molekülen würden wir auch noch die Spezifikation der Quantenzahlen mj und m'j verlangen, die die Orientierung der Reaktanden bzw. Produkte bezüglich einer gegebenen Richtung beschreiben (dies wird üblicherweise die Richtung der Relativgeschwindigkeit sein). Im allgemeinen wird man dazu Molekularstrahlen und Laser brauchen, wie wir an Beispielen sehen werden.
Das Idealexperiment müßte demnach den Streuquerschnitt und die Winkelverteilung der Produkte für einen derart vollständig spezifizierten Stossvorgang bestimmen. In der Praxis versuchen wir dem möglichst nahezukommen, nicht zuletzt, um mit der Theorie vergleichen zu können. Das bedeutet in der Reihenfolge immer größeren Details die Messung folgender Größen: Integraler Reaktionsquerschnitt bei vorgegebener Stossenergie, Winkelverteilungen der Produkte, möglichst zusätzlich die Geschwindigkeitsverteilung bei jedem Winkel, aus der man wegen der Energieerhaltung ihre innere Energie rekonstruieren kann, Zustandsanalyse der Produkte, Zustandsvorgabe der Reaktanden, Vorgabe der Orientierung der Reaktanden, Analyse der Orientierung der Produkte. Im übrigen sind alle diese Eigenschaften nicht unabhängig voneinander, so dass wir uns auch noch für die Korrelationen interessieren müssen!
Ein ideales Experiment, das alle diese Wünsche gleichzeitig erfüllt, hat es beisher noch nicht gegeben. Einzeln sind allerdings alle diese Daten schon einmal gemessen worden, auch Kombinationen von ihnen. Es ist klar, dass eine der ernsten Grenzen der Methode die Nachweisempfindlichkeit ist: Je genauer man das Ergebnis spezifiziert haben will, desto weniger Moleküle sind zum Nachweis da, und desto mehr geht das Signal im Rauschen des Detektors unter. Der Fortschritt in der Trennschärfe der Experimente der letzten Jahre wurde dabei von einer zweiten Entwicklung begleitet, nämlich von der wachsenden Komplexität der untersuchten Reaktionen. Laser, die bei den Experimenten mit einfachen Reaktanden die Auflösung erhöhen halfen, waren auch hier ein wesentliches Hilfsmittel des Fortschritts.
Natürlich verlangt man für große vielatomige Moleküle nicht das gleiche Detail wie für kleine. Hat man komplexe Edukte und Produkte mit vielen Freiheitsgraden, so führt die stärkere Summierung bzw. Mittelung über End- bzw. Anfangszustände zu Ergebnissen, die oft nicht weit von statistischen A-priori-Erwartungen entfernt liegen. Überdies verlaufen die Stöße großer Moleküle viel häufiger über einen langlebigen Stoßkomplex. Für größere Systeme haben wir daher bescheidenere Ziele, z.B. einfach nur zu wissen, was denn die primären Produkte überhaupt sind.
Als nächstes wollen wir einzelne experimentelle Methoden und typische
Resultate ansehen:
Chemilumineszenz
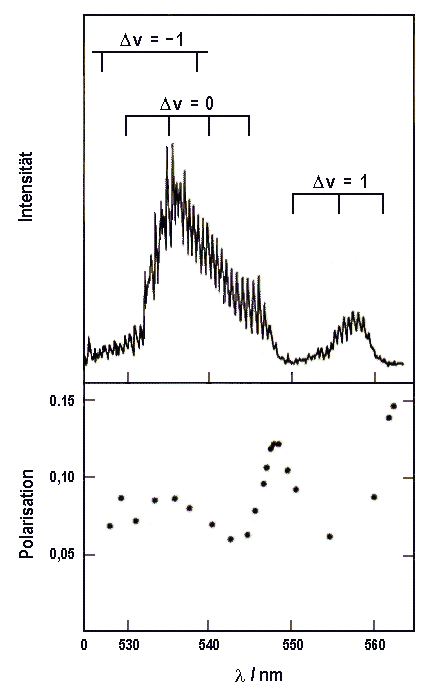 |
| Abb.
1: Chemielumineszenz von CaF(B 2S+)
aus der Reaktion von Ca mit F2.
Oben: Chemieluminszenz-Spektrum, das die Hauptsequenz
Δv
= 0 und die Nebensequenzen Δv
= ±1
zeigt. Unten: Polarisation der Chemielumineszenz, aus der man auf die Ausrichtung
des entstehenden CaF schließen kann.
(Nach M.G. Prisant, C.T. Rettner, R.N. Zare: J. Chem. Phys., 81, 2699 (1984)) |
Diese Methode besteht in der spektroskopische Analyse der Strahlung, die von angeregten Primärprodukten einer elementaren chemischen Reaktion ausgesandt wird, und hat das Ziel, die relativen Besetzungszahlen der angeregten inneren Zustände der Produktmoleküle zu messen. Besonders detaillierte Ergebnisse erhielt man für Reaktionen, in denen Halogenwasserstoff gebikdet wird, da dieser ein auflösbares Infrarotspektrum aussendet. Andere Experimente lieferten Schwingungsverteilungen in angeregten elektronischen Zuständen (*), indem deren sichtbares oder UV-Spektrum untersucht wurde, z.B.
Ca + F2 → CaF*(v', j') + F
Ein typisches Chemielumineszenzexperiment führt man in einem schnellen Flußreaktor unter stationären Bedingungen durch. Problematisch ist die Stossrelaxation (hauptsächlich der Rotationsverteilungen), deshalb muss man bei sehr niedrigen Drücken arbeiten. Üblicherweise kreuzt man zwei unkollimierte Düsenstrahlen und sorgt dafür, dass sie schnell abgepumpt werden. Die Strahlung aus der Reaktionszone wird gesammelt und in ein IR-Spektrometer fokussiert. Aus der Messung der relativen Linienintensitäten kann man bei bekannten Franck-Condon-Faktoren die relative Besetzung der Ausgangszustände errechnen. Wichtig ist, dass man die normalerweise sofort einsetzende schnelle Relaxation der Rotationsbewegung zu einen gegebenem Schwingungszustand "festsetzt", d.h. die relaxierten Moleküle an der Aussendung von Strahlung in den Detektor hindert. Extrapolation auf verschwindende Relaxation erlaubt dann die zuverlässige Bestimmung der Zustandsverteilung der frisch entstandenen Moleküle.
Die Beobachtung der Chemielumineszenz liefert relative Raten für die Bildung von Produktzuständen (v', j'). Absolutwerte können jedoch aus der Messung integraler Raten in konventionelle Experimente übernommen werden.
Beobachtet man elektronisch angeregte Produkte, so sind die Strahlungslebendsdauern
wesentlich kürzer als im Infraroten, was zu höheren Intensitäten
der Emission führt. Man kann dann nicht nur die Zustandsbesetzungen
messen, sondern oft weitere Details, wie die Polarisation der Strahlung,
die ihrerseits Aussagen über die Orientierung der Produkte macht.
Abbildung 1 zeigt ein Chemielumineszenzpektrum von CaF* aus der Reaktion
von Ca mit F2 mit Angabe der Schwingungsübergänge
und die Polarisation dieser Strahlung.
Der Laser als Pumpe und Analysator
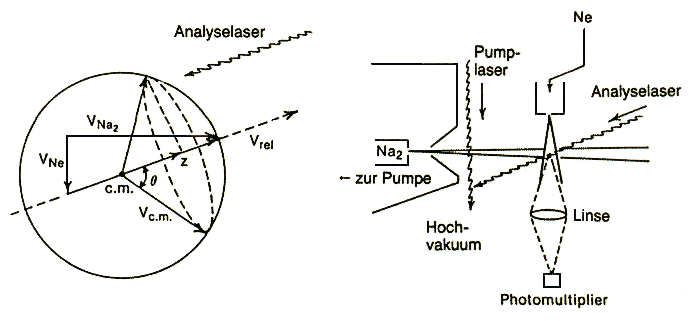 |
| Abb. 2: Schema einer Apparatur mit gekreuzten Molekularstrahlen und je einem Pump- und Analyse-Laser zur Messung zustandselektrischer Querschnitte. |
Die scharfe Frequenz, die hohe Leistung und die gute Kollimation des Lasers machen ihn zu einer idealen Photonenquelle, um Reaktanden zu präparieren oder Produkte zu analysieren. Eine typische experimentelle Anordnung für ein Molekularstrahlexperiment mit Lasern zeigt Abbildung 2. Sie enthält ein Paar gekreuzter Molekülstrahlen, von denen einer kurz vor der Stoßzone vom Pumplaser gekreuzt wird. Der hohe Photonenfluß dieses Lasers pumpt einen merklichen Anteil der Strahlmoleküle in einen gewünschten Quantenzustand. Die scharfe Frequenz sorgt dafür, dass dieser wohldefiniert ist, die Abstimmbarkeit (wenn vorhanden) für die Möglichkeit, verschiedene angeregte Zustände zu erreichen. Wenn das Laserlicht linear polarisiert ist, also der elektrische Feldvektor E eine feste Richtung (senkrecht zum Laserstrahl) hat, haben die Moleküle nach der Absorption im Laborsystem eine Ausrichtung: Die Absorption ist proportional zu |µE|², wo µ das molekulare Übergangsmoment (definiert im molekülfesten Koordinatensystem) und E die elektrische Feldstärke (definiert im Laborsystem) sind. Die Ausrichtung des Moleküls bestimmt sich relativ zu E und daher zu einem laborfesten Koordinatensystem.
Der Analyselaser fragt die Stoßprodukte ab. Die meisten Stöße führen in den elektronischen Grundzustand, regen jedoch verschiedene Rotations- und Schwingungszustände an. Eine wichtige Analysenmethode ist dann die laserinduzierte Fluoreszenz (LIF). Hier wird mit einem abstimmbaren Laser ein geeigneter Wellenlängenbereich im Sichtbaren und UV überstrichen, wodurch (bei passenden Wellenlängen) die Moleküle, deren Schwingungsverteilung man sucht, elektronisch angeregt werden. Ihre Fluoreszemz zurück in den elektronischen Grundzustand wird ohne spektrale Zerlegung gemessen. Da die Fluoreszenzlebensdauer elektronisch angeregter Moleküle kurz sind (im Nanosekundenbereich), haben sie sich während der Beobachtung praktisch nicht bewegt. Im Gegensatz dazu sind Fluoreszenzlebensdauern im Infrarot lang (> 1 ms), und die Moleküle könne während dieser Zeit das Beobachtungsgebiet leicht verlassen.
Auch der Analyselaser kann linear polarisiert sein. Beobachtet man die Doppler-Verschiebung des Übergangs, so kann man die Geschwindigkeitsverteilung des Produktflusses in Richtung des Laserstrahls messen. Liegt der Laserstrahl in Richtung der anfänglichen Relativgeschwindigkeit der Stoßpartner, v, so wird die Doppler-Verschiebung von der Komponente der Relativgeschwindigkeit nach dem Stoß w in Richtung v erzeugt. Mit anderen Worten, mit der Doppler-Verschiebung kann man bei bekannter Translationsenergie direkt die Winkelverteilung im Schwerpunktsystem bestimmen.
Photofragment-Spektroskopie
Dies ist eine Methode um die Dynamik der Photdissoziation zu studieren.
Was ist das Schiksal eines Moleküls nach der Absorption eines Photons,
dessen Energie die Schwelle für den Bruch einer oder mehrer chemischer
Bindungen übersteigt? In einfachsten Fall hat dabei das Molekül
einen elektronischen Übergang aus dem Grundzustand in einen abstoßenden
oder "anti-bindenden" elektronischen Zustand gemacht, der direkt zur Dissoziation
führt. Die Fragmente fliegen (im Schwerpunktsystem) in entgegengesetzter
Richtung und teilen die verfügbare Energie E = hn
- D0 zwischen sich auf. Ein Teil der Energie kann dabei
in innere Anregung der Fragmente gehen, d.h Rotations-, Schwingungs- oder
auch elektronische Anregung. Ein Beispiel ist
CH3I + hν(266 nm) → CH3 + I(2P)
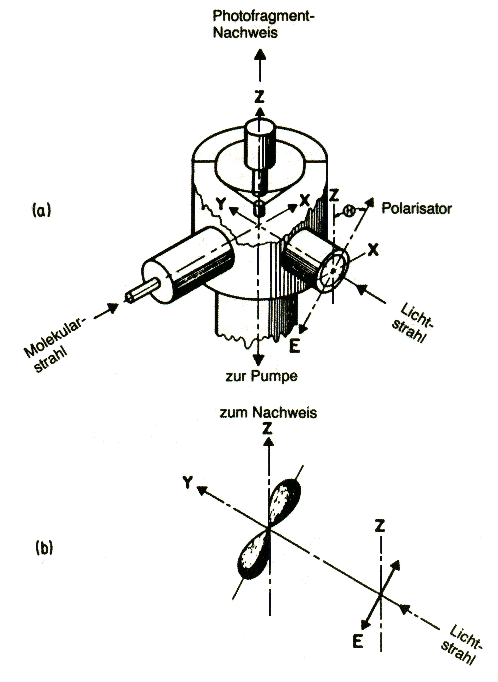 |
| Abb.
3: a) Photofragment-Spektrometer. Ein Molekularstrahl in X-Richtung
wird von einem Lichtstrahl in Y-Richtung gekreuzt, die Bruchstücke
aus der Photodissoziation, die in die Z-Richtung fliegen, werden massenspektrometrisch
nachgewiesen. Das Licht wird gepulst, so dass die Geschwindigkeit der Fragmente
und damit ihre Translationsenergie bestimmt werden kann. Der Polarisator
erlaubt die Untersuchung der Ausbeute der Photodissiziation als Funktion
des Winkels Q
zwischen dem elektrischen Lichtvektor E und der Richtung des Übergangsdipols
µ des absorbierenden Moleküls.
b) Winkelverteilung der wegfliegenden Photofragmente. Wenn das zweiatomige Molekül langsam rotiert, erreichen nur diejenigen Moleküle den Detektor, die in Richtung der Z-Achse zerfallen sind. Die Absorptionswahrscheinlichkeit des Moleküls ist ~ |µ·E|². Ist µ entlang der Molekülachse gerichtet und der Detektor in Z-Richtung, so ist |µ·E|² ~ cos²Q. Diese Verteilung, die Zylindersymmetrie um E hat, ist ebenfalls angedeutet. |
wo ein großer Bruchteil der I-Atome in den angeregten 2P1/2-Zustand geht, und außerdem im CH3-Radikal die "Regenschirm"-Schwingung angeregt ist und im Infraroten fluoresziert. Der Rest der verfügbaren Energie geht als Rückstoß in die Translation. Es ist ein wichtiges Ziel, diese Aufteilung der Energie zu messen. Das kann z.B. geschehen, indem man die Energieverteilung der Translation eines der Fragmente mißt. Höhere Auflösung bekommt man, wenn man die Besetzung der inneren Zustände der Fragmente direkt misst, gewöhnlich mittels LIF. Ein weiteres Ziel wäre die Bestimmung der Richtung des Übergangsmomentes relativ zur Molekülachse.
In einem typischen Photofragmentexperiment wird ein Molekülstrahl
von einem intensiven, gepulsten, polarisierten Laserstrahl gekreuzt (Abbildung
3). Die Polarisationsebene kann gedreht werden, so dass sich der Winkel
zwischen dem E-Vektor des Lichts und der Flugrichtung der Fragmente (nachgewiesen
z.B. durch ein Massenspektrometer) ändern läßt. Die so
gemessene Winkelverteilung der Produkte gibt direkt Auskunft über
den Winkel zwischen Bindungsachse und Übergansdipolmoment.
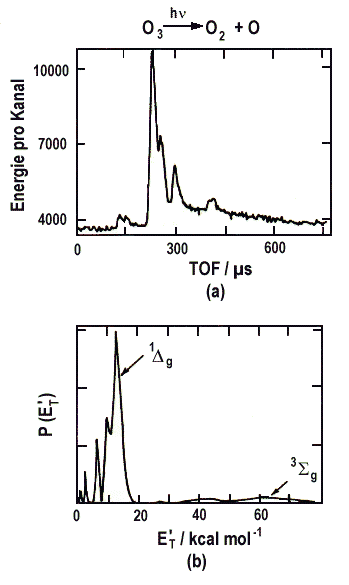 |
| Abb.
4: a) Flugzeitverteilung des Bruchstücks O2
aus der Photodissoziation von O3
bei 266 nm. Der Nachweis erfolgt 30° neben dem Laserstrahl. b) Dieselben
Daten, aber jetzt als Verteilung der Translationsenergie im Schwerpunkt-
system, P(E't). Das höchste Maximum entspricht der Bildung
von O(1D)+O2(1Dg,v'=
0). Daneben sind Maxima zu v = 1, 2 und 3 zu sehen. Der schwache hochenergetische
"Schwanz" gehört zum Zerfall in O(3P)+O2(3Sg-).
Eine genaue Analyse zeigt, dass die relativen Ausbeuten der Schwingungszustände
0, 1, 2 und 3 sich wie 57:24:12:7 verhalten, und dass rund 17% der übrigen
Energie in Rotationsanregung des O2
gehen.
(Nach R.K. Sparks, L.R. Carlson, K. Shobatake, M.L. Kowalzcyk, Y.T. Lee: J. Chem. Phys., 72, 1401 (1980)) |
Um die Translationsenergie zu bestimmen, triggert der Lichtimpuls den Detektor, der die Zahl der Fragmente pro Zeit in Abhängigkeit von der Verzögerung zählt. Die Verteilung der Ankunftszeit wird in eine solche der Translationsenergie umgerechnet. Da die verfügbare Energie bekannt ist, kann man die innere Energie der Fragmente angeben; es ist
Pi(E'i) = Pt(E − E'i) = Pt(E't)
wo Pt(E't)dE't der Bruchteil der Produkte mit Translationsenergie zwischen E't und E't + dE't ist. Benutzt man nciht nur einen polarisierten Pumplaser, sondern auch einen polarisierten Anaylselaser, so kann man neben der Energieverteilung auch noch die relative Orientierung der Fragmente nach dem Stoß messen.
Ein Beispiel, wo man aus der Messung der Rückstoßgeschwindigkeit den Schwingungszustand der Produktmoleküle bestimmen konnte, ist die Photodissoziation von O3:
O3 → O2 + O
Die in Abbildung 4 gezeigte Verteilung der inneren Energie des O2 zeigt, dass diese weit über dem thermischen Gleichgewicht liegt. Auch diese spezielle Energieverteilung zeigt den direkten Charakter dieses Dissoziationsvorgangs. Die Besetzungsinversion in den Produkten solcher Prozesse ist häufig in chemischen Lasern verwandt worden.
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.