Die Dynamik einer chemischen Reaktion ist vollständig bestimmt
durch die Zustände der Edukte und die Hyperfläche der potentielle
Energie, auf der die Reaktion abläuft. Diese Hyperfläche bestimmt
sowohl welche innere Anregung die Edukte haben müssen, um die Reaktion
zu beschleunigen, als auch in welchen Zuständen die Produkte gebildet
werden; ob die freiwerdende Energie mehr in die innere Anregung (Rotation,
Vibration oder elektrisch angeregte Zustände) oder mehr in die Translation
umgesetzt wird. Um ein solches zwischenatomares Potential zu kennen, müssen
wir wissen, wie sich die Wechselwirkung als Funktion der Konfiguration
des Gesamtsystems - also vom Übergang der Reaktanten zu den Produkten
- verhält.
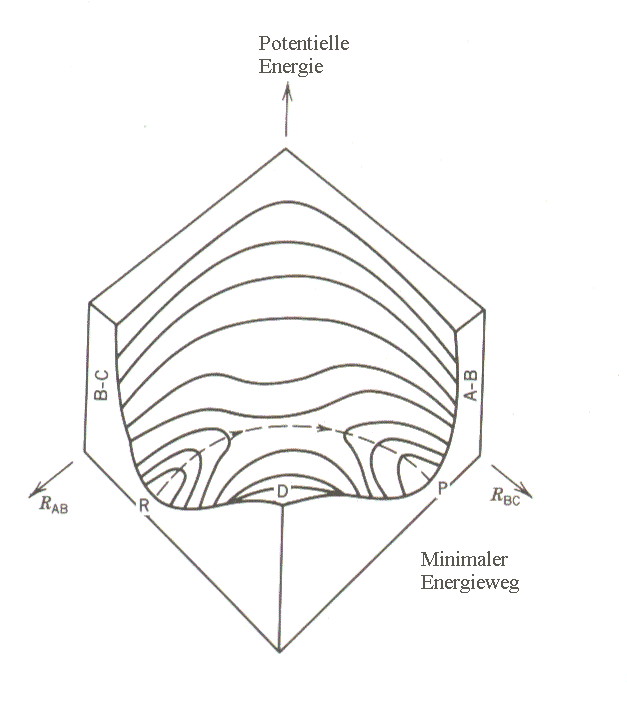 |
| Abb. 1: Potentielle Energie für die Reaktion A + BC → AB + C für eine lineare Reaktionsgeometrie. |
A + BC → AB + C
lässt eine grafische Darstellung der potentiellen Energie nicht
mehr zu, es sein denn, man betrachtet eine unter einem bestimmten Winkel
bestehende (A-B-C) Anordnung, bei der nur zwei unabhängige zwischenatomare
Abstände berücksichtigt werden müssen. In diesem Fall kann
man das Potential als Funktion zweier Koordinaten (Abstand des Atoms A
zum Atom B im BC-Eduktmolekül und Abstand des Produktatoms C zum Atom
B im Produktmolekül AB) in einer Konturenkarte darstellen, in der
Equipotentiallinien, ähnlich den Höhenlinien einer topografischen
Karte, eingezeichnet sind. Ein Beispiel einer solchen Darstellung der potentiellen
Energie ist in Abbildung 2 für das H+H2-System gegeben.
Die ausgezogenen Linien sind Linien gleicher potentieller Energie, die
gestrichelte Linie gibt den Weg der minimal benötigten Energie an,
um vom Tal der Edukte (A, BC) zum Tal der Produkte (AB, C) zu gelangen.
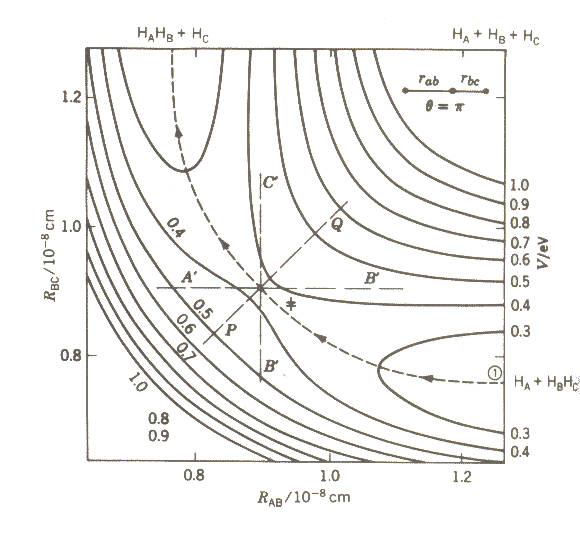 |
| Abb. 2: Potentielle Energie für die Reaktion HA + HBHC→ HAHB + HC (lineare Reaktionsgeometrie) |
Die Energiedifferenz zwischen dem höchsten Punkt auf der gestrichelten Kurve und den völlig getrennten Reaktanten kann man als klassische Energiebarriere für diese Reaktion ansehen. "Klassisch", weil Quanteneffekte bei der Bewegung der Atome unter dem Einfluß des Potentials bisher nicht berücksichtigt wurden. Es geht z.B. für rAB ® ¥ die potentielle Energie in die des ungestörten Moleküls BC über. Löst man nun die Schrödingergleichung für das Molekül, dann erhält man die Vibrations- und Rotationszustände von BC, wobei der niedrigste Zustand natürlich oberhalb des Minimums der Potentialkurve liegt. Ähnliche Quanteneffekte gelten aber überall auf der Hyperfläche, die Größe der Nullpunkt-Energie ist jedoch nicht mehr fest.
Um besser zu verstehen, wie die energetische Schwelle für eine
Reaktion zustandekommt, wollen wir den Ebergieaufwand bei der Bewegung
aus dem Gebiet der Reaktanden in das der Produkte betrachten. Wir definieren
den Reaktionsweg als den Weg der minimalen Energie, der aus dem
Tal der Reaktanden in das der Produkte führt.
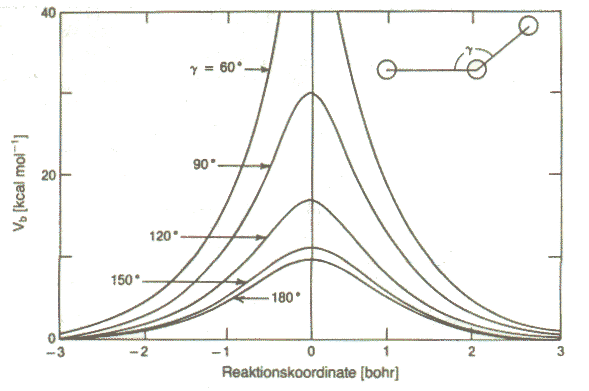 |
| Abb. 3: Potentialbarrieren für dir Reaktion H + H2 bei verschiedenen Bindungswinkeln als Funktion der Reaktionskordinate auf der LSTH-Fläche (s. Abb. 2). Das globale Minimum für die Barriere ist bei g = 180° und hat den Wert 9,8 kcal·mol-1. |
Abbildung 3 zeigt ihn für das H3-System, bei dem die kollineare Schwelle diejenige mit der niedrigsten Energie ist (≈ 40 kJ· mol−1 ≈ 1,7 eV): Die Energie, die nötig ist, um ein H-Atom in einer konzertierten Bewegung auszutauschen, ist weniger als 30 % der Dissoziationsenergie des H2. Die Existenz solcher niedrigen "Pässe" zwischen den Tälern von Reaktanden und Produkten ist es, die zur Bevorzugung des konzertierten, bimolekularen Mechanismus beim Austausch von Atomen oder Atomgruppen führt.
Da der Reaktionsweg jeweils das "Tal" der Fläche benutzt, nimmt die potentielle Energie bei seitlicher Abweichung von diesem Weg zu. In der Nähe der Barriere hat das Potential daher die Form eines Sattels. Der Ort der Barriere wird daher auch als Sattelpunkt der Fläche bezeichnet. Auch die Krümmumg des Potentials quer zum Reaktionsweg ändert sich entlang desselben. Die zugehörige Kraftkonstante geht dabei (im Falle A + BC → AB + C) von derjenigen des Reaktanden BC stetig in die des Produktes AB über. Auf dem Sattel ist es die Kraftkonstante der symmetrischen Streckschwingung von ABC. Die asymmetrische Streck"schwingung" an dieser Stelle ist die Bewegung auf dem Reaktionsweg selbst.
Neben dieser allgemeinen theoretischen Betrachtung haben Polanyi und seine Mitarbeiter untersucht, wie sich die Beschaffenheit der potentiellen Hyperfläche auf die Form der freiwerdenen Energie auswirkt und welche Form der Anregung die Reaktanten besitzen müssen, um eine Reaktion zu beschleunugen oder zu verlangsamen. Eine solche Betrachtungsweise erlaubt es, qualitativ Reaktionsprozesse zu studieren, und es besteht die Möglichkeit, sie auf allgemeinere Systeme wie A + BCD → AB + CD zu übertragen.
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.