Von der Potentialfläche zur Dynamik
Schon in den dreißiger Jahren erkannte man, dass es einen direkten
Weg von der Potentialfläche zur Stoßdynamik gibt: Die numerische
Lösung der klassischen Bewegungsgleichungen für die Atome. Sie
erlaubt es, alle wesentlichen Erscheinungen der Reaktionsdynamik zu simulieren,
soweit nicht Quanteneffekte beteiligt sind. Man macht diese rechnerischen
Untersuchungen aus zwei Gründen, (1) zur Diagnose allgemeiner Trends,
d.h. dynamische Erscheinungen, die mit verschiedenen topografischen Eigenschaften
der Fläche oder mit Parametern des Systems wie Stoßenergie,
Massen u.a. zusammenhängen, und (2) zur Untersuchung einzelner Reaktionen
in der Hoffnung, beobachtete Eigenschaften auf diese Weise erklären
zu können.
Grundsätzlich besteht das Verfahren darin, ein Potenzial vorzugeben,
einen Satz von Anfangsbedingungen auszuwählen und dann die klassischen
Bewegungsgleichungen für die drei (oder mehr) Teilchen zu lösen.
Für jede Anfangsbedingung wird die zeitliche Entwicklung der Koordinaten
jedes Teilchens im Schwerpunktsystem berechnet. Das Ergebnis ist eine Trajektorie
des Systems, die in verschiedener Weise grafisch dargestellt werden kann.
Sehr aussagekräftig ist z.B. die Darstellung der Abstände Rij(t)
aller Paare (i, j) von Atomen. Für ein Dreiteilchensystem sind die
Kurven leicht verständlich (Abb.1).
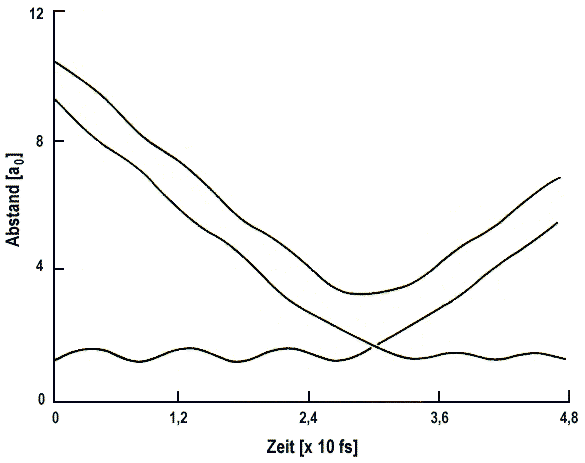 |
| Abb. 1a: Klassische Trajektorie für Stöße von H mit D2 (v = 0) bei Et = 79 kcal·mol-1. Die oberste Kurve führt nicht zur Rektion, während die andere Kurve reaktiv ist und zu HD + D führt. |
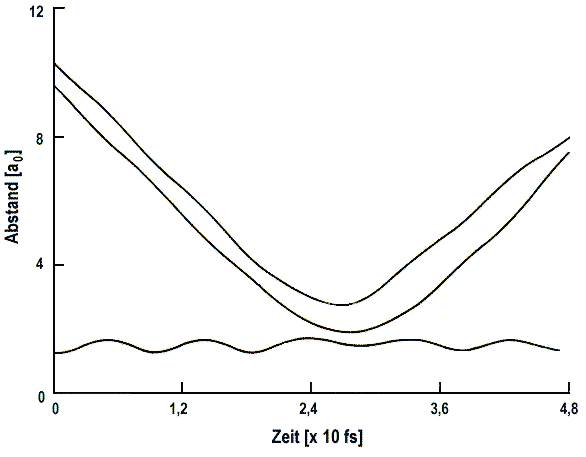 |
| Abb. 1b: Klassische Trajektorie für Stöße von H mit D2 (v = 0) bei Et = 79 kcal·mol-1, die nicht zur Reaktion führen. |
Jede Trajektorie in Abbildung 1 gehört zu einer speziellen Wahl der Anfangskoordinaten und -impulse der Teilchen. Um physikalisch interessante Eigenschaften wie Streuquerschnitte, Reaktionsraten o.ä. zu bekommen, muss man über einige oder alle Anfangsbedingungen mitteln. Selbst um solche Rechengrößen wie die Reaktionswahrscheinlichkeit P(b) bei gegebener Energie zu bestimmen, muss man ja über alle möglichen Anfangsorientierungen der Reaktanden bei festen b mitteln. Im Prinzip (und manchmal sogar in der Praxis) kann der Reaktionsquerschnitt für orientierte Reaktanden allerdings gemessen werden. Dann darf die entsprechende Trajektorienrechnung natürlich nicht über die Orientierung gemittelt werden, sondern muss den Querschnitt als Funktion dieses Winkels berechnen.
Es gibt jedoch einen anderen, tieferen Grund, warum es unausweichlich ist, über Anfangsbedingungen zu mitteln, ein Grund, der selbst beim Vergleich mit einem idealen Experiment bestehen bleibt. Ein wesentlicher Unterschied zwischen klassischer und Quantenmechanik ist nämlich die Zahl der Anfangsbedingungen, die man für eine bestimmte Trajektorie überhaupt angeben darf. In der klassischen Mechanik muss man für jeden Freiheitsgrad Ort und Impuls festlegen. In der Quantenmechanik folgt jedoch aus der Unbestimmbarkeitsrelation, dass, wenn z.B. der Impuls genau festgelegt wird, die Ortkoordinate beliebig unbestimmt ist. Da Moleküle nun einmal Quantensysteme sind, besteht eine vollständige Festlegung von Anfangsbedingungen für einen Stoss bei einem System von n Freiheitsgraden aus n Quantenzahlen. Im Gegensatz dazu verlangt eine klassische Trajektorie für das System 2n Anfangsbedingungen. Die Methode der klassischen Trajektorie simuliert dieses Quantenverhalten, indem viele klassische Trajektorien gestartet werden (dieselben wie im Quantenfall), während die anderen n variiert werden. Das Endergebnis bekommt man durch Mittelung über diese variierten Anfangsbedingungen. Im einfachsten Fall einer Reaktion A + BC muss man über die Phase der Schwingung und Rotation und die Drehimpulsrichtung von BC mitteln.
Die Auswahl mder Anfangsbedingungen mit dem Ziel, hinterher darüber zu mitteln, wird oft unter dem Namen Monte-Carlo-Verfahren in der Form einer Zufallsauswahl durchgeführt. Daher nennt man häufig das ganze Verfahren der Berechnung beobachtbarer dynamischer Eigenschaften durch Mittelung über klassischen Trajektorien die Monte-Carlo-Methode. Daneben gibt es auch systematische (d.h. nicht zufällige) Verfahren, die Anfangsbedingunen auszuwählen. Diese können unter Umständen zur Berechnung von Mittelwerten bei vorgegebenem Fehler effizienter als die Monte-Carlo-Methode sein. Je nachdem bis zu welchem Detail man die beobachtbare Größe festlegen will, braucht man eine oder weniger große Zahl von Trajektorien, um eine bestimmte Genauigkeit zu erreichen. Typisch sind 103 Trajektorien für grobe Daten, z.B. integrale Querschnitte, während doppelt differentielle Querschnitte eher 106 verlangen (z.B. Kenntnis der Vibration und Rotation des Produktmoleküls).
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.