Bei der Beschreibung von Festkörpern gehen wir stets von der wohlgeordneten Struktur eines idealen Kristalls aus. Die potentiellen Energien sind weit höher als die kinetischen Energien. Bei Gasen ist unser Startpunkt die völlig ungeordnete Bewegung der Moleküle. Hier überwiegen die kinetischen Energien die potentiellen. Flüssigkeiten liegen zwischen diesen beiden Extremen: Es gibt noch eine gewisse Nahordnung, aber kaum eine Fernordnung.
Die Teilchen in einer Flüssigkeit werden durch zwischenmolekulare
Kräfte zusammengehalten, ihre kinetischen Energien sind jedoch vergleichbar
mit ihren potentiellen Energien. Die ganze Struktur ist daher sehr beweglich.
Für eine mathematische Beschreibung müssen wir uns mit den mittleren
Positionen der Teilchen begnügen. Die radiale Verteilungsfunktion
g ist so definiert, dass g(r)r2dr die Wahrscheinlichkeit
angibt, in einem Intervall dr im Abstand r von einem Teilchen ein zweites
Teilchens anzutreffen.
Für einen idealen Kristall besteht g(r) aus einer Abfolge von
scharfen Peaks, die die festen Positionen der Atome im Kristall angeben.
Diese Regelmäßigkeit setzt sich bis an die Grenzen des Kristalls
fort, wir sprechen daher von einer Fernordnung der Atome im Kristall.
Beim Schmelzen des Kristalls geht diese Fernordnung verloren, und wenn
wir in großer Entfernung von einem Teilchen nach der Wahrscheinlichkeit
schauen, ein zweites Teilchen anzutreffen, so erhalten wir einen konstanten
Wert. In direkter Umgebung eines Teilchens können die nächsten
Nachbarn jedoch immer noch ungefähr an ihren alten Positionen sitzen,
und selbst wenn sie durch neue Atome ersetzt werden, werden diese mit einiger
Wahrscheinlichkeit die Positionen der alten besetzen. Wir können daher
auch in der Flüssigkeit noch eine Schale von nächsten Nachbarn
eines Atoms bei einem Abstand r1 finden, und in etwas größerer
Entfernung vielleicht auch noch eine Schale mit übernächsten
Nachbarn bei einem Abstand r2. Diese Erscheinung bezeichnen
wir als Nahordnung. Für die radiale Verteilungsfunktion bedeutet
das, dass sie bei kurzen Abständen oszilliert, bei r1
ein Maximum besitzt, bei r2 ein zweites, aber weniger ausgeprägtes
Maximum, vielleicht noch weitere bei größeren Abständen,
und dann für sehr große Abstände gegen einen konstanten
Wert geht.
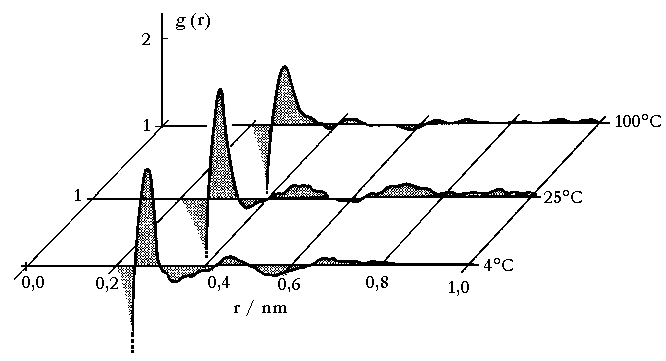 |
| Abb. 1: Die radiale Verteilungsfunktion der Sauerstoffatome in flüssigem Wasser bei drei Temperaturen. Mit steigender Temperatur verschieben sich die Maxima nach außen. |
Die Berechnung von g aus dem zwischenmolekularen Potential kann auf
verschiedene Weisen erfolgen. Numerische Verfahren nehmen 1000 Teilchen
in einem bestimmten Volumen (je schneller die Computer, desto größer
die Zahl), deren Bewegungen verfolgt werden, und simulieren den Rest der
Flüssigkeit durch identische Kopien dieses kleinen Behälters,
die um ihn herum angebracht werden.
Ein häufig angewendetes Verfahren zur Simulation von Flüssigkeiten
ist das Monte-Carlo-Verfahren. Dabei werden die Moleküle um
kleine, aber völlig zufällige Strecken "verschoben". Ob die entstandene
neue Konfiguration akzeptiert wird, wird dann nach bestimmten Regeln entschieden:
Man kann zeigen, dass dieses Verfahren im Gleichgewicht für eine beliebige Konfiguration mit der Energie EN eine Wahrscheinlichkeit proportional zu e-ΔEN/kT liefert, im Einklang mit der Boltzmann-Verteilung. In den so erzeugten Konfigurationen werden dann einfach die Abstände aller Paare von Teilchen vermessen und diese über viele Konfigurationen gemittelt.
Ein anderer Ansatz zur Simulation von Flüssigkeitsstrukturen ist die Molekulardynamik. Hierbei berechnet man ausgehend von einer zufälligen Anfangsverteilung die Bahnen aller Teilchen unter dem Einfluss der zwischenmolekularen Potentiale. Dazu werden die Newtonschen Bewegungsgleichungen in kleinen Zeitschritten (etwa 10-15s, kürzer als das mittlere Intervall zwischen zwei Stößen) numerisch integriert. Eine Berechnung wird für Tausende derartiger Zeitschritte durchgeführt, und für jede dabei durchlaufende Konfiguration werden die Abstände zwischen den Teilchen ausgewertet.
Aus der radialen Verteilungsfunktion können wir die thermodynamischen Eigenschaften der Flüssigkeit berechnen. Beispielsweise ist der Beitrag der Paarwechselwirkungen zur Inneren Energie durch das Integral
gegeben. Das bedeutet, der Beitrag ist gleich der mittleren Wechselwirkung Ew zweier Teilchen, gewichtet mit einem Faktor g(r)r2dr, der die Wahrscheinlichkeit angibt, mit der ein Abstand zwischen r und r+dr auftritt. Entsprechend ist der Beitrag der Paarwechselwirkungen zum Druck durch
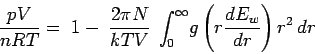
gegeben. Die Größe r · dEW/dr wird als Virial bezeichnet, die angegebene Gleichung daher auch als Virialzustandsgleichung.
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.