The Harmonic Oscillator
 |
The potential energy of a diatomic molecule has a minimum energy when the atoms are separated by some defined distance. The potential energy V(x) expansion in series relative to state of equilibrium x0(for which dV(x)/dx|x0 = 0 ) gives V(x) = V(0) + ½ d²V(x)/dx²|x0 x2. If we choose the zero point of energy V(0) and assign d²V(x)/dx²|x0 as constant k then we obtain the potential approximation
V(x) = ½ kx² (x is a value that corresponds to deviation from equilibrium position)
The Schroedinger equation
(−h²/2md²/dx² +
½ kx²) y = E y
Since A and B vibrate in the opposite directions then we will change m to
reduced mass µ = mAmB/mA+mB. If we introduce a new distance variable
quantity z² = x² (kµ/h²)½ then we finally obtain the Schroedinger
equation:
(−d²/dz² +
z²) y(z) = 2
E/hw·y(z)
where the vibration frequency is as follows w = (k/µ)½ . A reader who is interested in more detailed mathematical solution can find it here.
Generally it's considered for possible energy levels:
| En = |
where n can take the following values n = 0, 1, 2, .... .
The energy ground state Eo = ½ hw is assigned as the zero point energy. The wavefunction of
this lowest energy state is the Gauss function:
The highest possibility of the system being in the ground state can be found at z=0, or equilibrium. In general one can obtain for the wavefunction of
n-state
| yn(z) = Cn Hn(z) e−z²/2 |
where constants Cn can be obtained from the normalization
condition -¥ò+∞yn*
yn dz = 1 in the following way:
| Cn = 1/(π½n!2n )½ |
and the functions Hn(z) are the Hermitian
polynomials:
| H0(z) = 1 |
| H1(z) = 2z |
| H2(z) = 4z2 − 2 |
| H3(z) = 8z3 − 12z |
| H4(z) = 16z4 − 48z2 + 12 |
| H5(z) = 32z5 − 160z3 + 120z |
| H6(z) = 64z6 - 480z4 + 720z2 − 120 |
 |
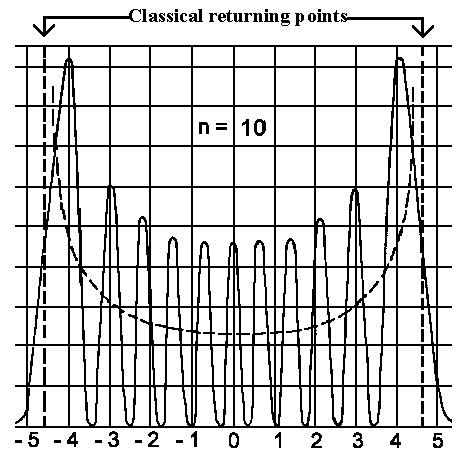 |
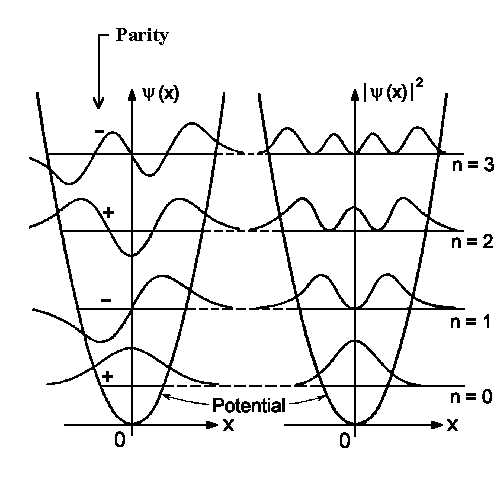
| The wavefunction (to the left) and the detection probability (to the right) for four lowest vibrational states. | The detection probability for the vibrational state n=10. The dashed line gives us the classical detection probability. |
The highest probability of locating the particle in the ground state is in the equilibrium
position. The quantity |yn|2
would be higher in the classical turning points region E = V(x).
The
dependence of classical oscillator detection probability is given by 1/w(xm²−x²)½. This
curve (see dashed line on the right illustration) is assigned to probability
density in the range [x, x+dx]; i.e. the highest chance is in the classical
turning points xm. One can find that quantum description converges with the
classical description for higher states.
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.