Allgemein läßt sich jede Funktion in eine Reihe nach einen
vollständigen Satz von Funktionen entwickeln:
| y(x) = åi ci φi(x) |
Zur Bestimmung aller Koeffizienten ci in der Reihenentwicklung wird von links mit Fk* multipliziert und über den gesamten Raum integriert.
ò fk*(x) y(x) dx = åi ci òfk*φidx = ck
Der sehr abstrakte Entwicklungssatz, dass sich jede Funktion in eine Reihe mit einem vollständigen Satz von Funktion entwickeln läßt, ist von fundamentaler Bedeutung für die Chemie.
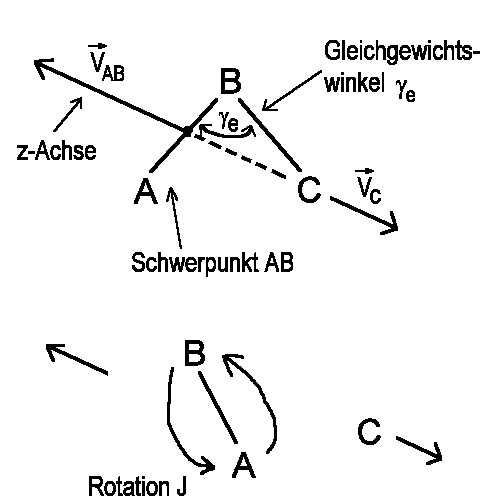 |
| Abb.1: Einfacher Bindungsbruch eines dreiatomigen Moleküls, ABC. |
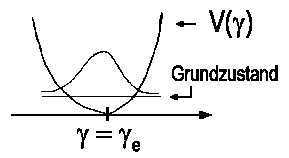 |
| Abb.2: Schwingungsgrundzustand (Biegeschwingung) des dreiatomigen Muttermoleküls ABC |
Betrachten wir z. B. den Zerfall eines dreiatomigen Moleküls ABC
in das Molekül AB und das Atom C, wobei die ursprüngliche Bindung
B-C einfach "wegfallen" soll, d. h. man stelle sich anschaulich B-C durch
eine "Stange" fest verbunden vor, die dann entfernt werden soll. Ohne auf
eine detaillierte quantenmechanische Beschreibung einzugehen, kann man
sich vorstellen, dass die Wellenfunktion, die den Grundzustand des
Muttermoleküls (also die Situation vor dem Zerfall) beschreibt, sich
in den Produkten wiederfinden muss, da sich ja bis auf eine Veränderung
der Ortskoordinate R, die den relativen Abstand der beiden Produkte beschreibt,
nichts geändert hat (Abb. 1).
Wir stellen also die ursprüngliche Wellenfunktion des Grundzustandes
y(G)
(Abb. 2) durch Terme der Produktwellenfunktion φ(P)
dar:
y(G) = åk ck φ(P)
ck = ò fk(P)* y(G) dV
Diese Berechnung, übertragen auf den Zerfallsprozeß, bei
dem das Produkt AB rotieren soll, bedeutet nichts anderes als die Grundzustandswellenfunktion
y(G)nach
den Rotationseigenfunktionen zu entwickeln. Da der Zerfall in der Ebene
stattfinden soll, sind die Rotoreigenfunktion unsere Kugelflächenfunktionen
Yl,m=0(J,j).Die Funktion y(G)
soll nun nur den für den Zerfall wichtigen Teil, nämlich die
Knickschwingung um den Gleichgewichtswinkel
γe,
beschreiben.
Für den niedrigsten Schwingungszustand wird die Wellenfunktion
durch eine Gaußkurve y(G)(γ)
~ exp[-α(γ-γe)2]
beschrieben (s. Abbildung), wie wir es aus der Behandlung des harmonischen
Oszillators kennen. Wir wollen nun noch die Nomenklatur für die
speziellen Yl,m=0 hier ändern und sie YJ(γ)
nennen; dann sind die Entwicklungskoeffizienten gegeben durch :
cJ = ò YJ(γ) exp[-α(γ-γe)2] sinγ dγ (sinγ dγ = dV)
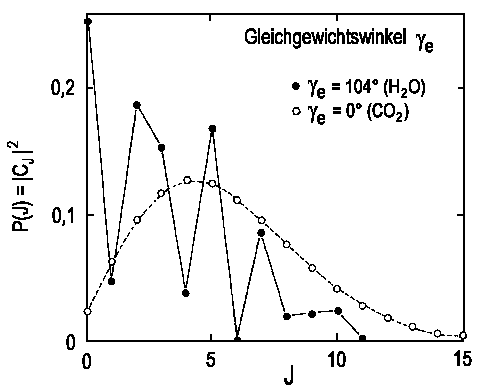
Die Wahrscheinlichkeit, eine bestimmte Rotation vorzufinden, ist gegeben durch P(J)=|cJ|2.
In der unteren Abbildung sind die so berechneten Verteilungen für H2O ® OH(J)+H und CO2® CO(J)+O angegeben. Die dazu benötigten Integrale sind lösbar, man muss lediglich γe und a kennen, die vom jeweiligen Molekül abhängen (Gleichgewichtswinkel γe ist über die Molekülstruktur gegeben und αläßt sich über die Knickschwingungsfrequenz berechnen).
 |
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.