In general, it is possible to replace any wave function by a complete series of eigenfunctions:
|
ψ(x) = Σi ci φi(x)
(all φi shall be orthonormal) |
For determination of all coefficients ci in the expansion series, the equation above is multiplied from the left side with φk* and then integrated within space.
∫ φk*(x)·ψ(x)dx = ∑i ci ∫φk*·φi dx = ck
Wave function expansion, a very abstract approach, is of fundamental importance in chemistry.
 |
 |
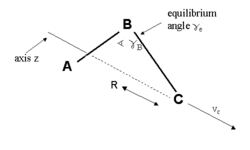
| Fig.1: Simple breakage of bond within the three atomic molecule ABC |
Thus, we replace the original wave function for the ground state
The coefficients ck are calculated using
This calculation, transferred to the process of the molecule's decomposition, is identical to an approach where the ground state wave function Ψ(G) is obtained by expanding the rotational eigenfunctions. As decomposition shall take place in a planar system, the spheric surface function Yl,m=0(θ,φ) represents the rotational eigenfunctions. Function ψ(G) is confined to the aspect relevant for decomposition, i.e. the bending mode vibration around the angle of equilibrium γe.
 |
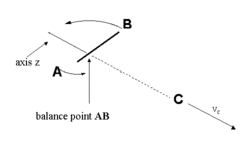
| Fig.2: Normal mode of vibration (bending) of the parent molecule ABC |
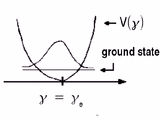
In analogy with our mathematical treatment of the harmonic oscillator in its lowest vibrational state, the wave function of interest has gaussian characteristics (comp. Fig.2).
Having replaced Yl,m =0 by YJ(γ); the expansion coefficients are obtained from:
with sin γdγ = dV.
The probability to observe are certain rotational state is given by
The diagram below depicts the calculated distribution for H2O → OH(J) + H and CO2 → CO(J)+O. It is possible to solve the necessary integrals for known values of &gammaeα. Note that the molecular structure defines the equilibrium angle γe and the value for α can be calculated from the frequency found for the bending mode of molecular vibration.
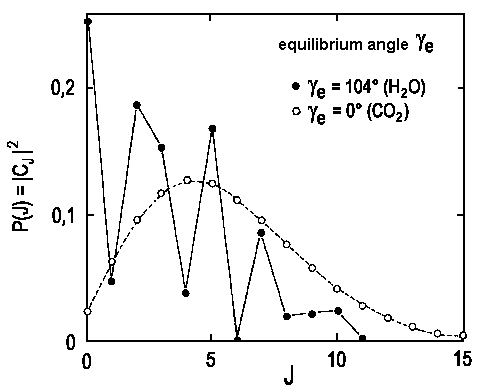 |
| Fig.3: The relative number of product molecules in the rotational states J. The theoretical treatment assumes that the ground state vibrational energy is completely transferred in rotational energy of the diatomic product. (filled dots for OH, empty dots for CO). |
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.