The corresponding vibrational energy states are the energies of the harmonic oscillator:
In general, when the approximation in eq. (55) is not valid, the energy levels
should be obtained as a solution of the corresponding Schrödinger equation, however, they
still can be classified by the quantum number ![]() (for a diatomic molecule case).
(for a diatomic molecule case).
If the molecule in its equilibrium position has a dipole moment, as is always the case for the heteroatomic molecules, this dipole moment will in general change if the internuclear distance changed. Thus, on the basic of classical electrodynamics the molecular vibration would lead to the emission of light at the oscillation frequency. Conversely, the oscillator could be set in vibration by absorption of light at this frequency. Therefore, all heteroatomic molecules in principle are said to be infrared active, that is they can absorb or emit infrared radiation. Contrary, all homoatomic diatomic molecules do not have any dipole moment and cannot set in vibration by absorption the infrared light. These molecules are said to be infrared inactive.
The selection rules for the vibrational transitions in a harmonic oscillator-like molecule are
As the energy difference between each two neighbor vibrational energy levels is ![]() (see eq. (56)), the vibrational spectrum would contain only one line
which is in fact detected experimentally. This line is called fundamental line. The
wavelength of this line for different diatomic molecules usually lies in near IR spectral range
(see eq. (56)), the vibrational spectrum would contain only one line
which is in fact detected experimentally. This line is called fundamental line. The
wavelength of this line for different diatomic molecules usually lies in near IR spectral range
![]() .
.
However, for high lying vibrational energy states the harmonic oscillator approximation in is
not valid any more. Usually additional terms proportional to ![]() ,
, ![]() has to be
added to the expression for the potential curve in eq. (55) which leads to the case
of the anharmonic oscillator. For anharmonic oscillator the selection rule in
eq. (57) is not valid and additional lines appear in the molecular vibration
spectra corresponding to transitions with
has to be
added to the expression for the potential curve in eq. (55) which leads to the case
of the anharmonic oscillator. For anharmonic oscillator the selection rule in
eq. (57) is not valid and additional lines appear in the molecular vibration
spectra corresponding to transitions with
![]() , and so on. These
transitions are called second harmonic, third harmonic, and so on. The
intensity of the harmonic transitions transitions is usually much smaller than the intensity of
the fundamental line.
, and so on. These
transitions are called second harmonic, third harmonic, and so on. The
intensity of the harmonic transitions transitions is usually much smaller than the intensity of
the fundamental line.
In high resolution spectra the vibrational lines in the near IR are resolved into a number of
individual lines which are due to vibrational-rotational transitions. A detailed
quantum mechanical analysis of simultaneous vibrational and rotational transitions shows that
the rotational quantum number ![]() changes by
changes by ![]() during the vibrational transition. If the
molecule also possesses angular momentum about its axis, (for instance,
during the vibrational transition. If the
molecule also possesses angular momentum about its axis, (for instance, ![]() ), then the
selection rule also allows
), then the
selection rule also allows ![]() . The appearance of the vibrational-rotational spectrum
of a diatomic molecule can be discusses in terms of the combined vibrational-rotational terms
. The appearance of the vibrational-rotational spectrum
of a diatomic molecule can be discusses in terms of the combined vibrational-rotational terms
When the vibrational transition
![]() occurs
occurs ![]() changes by
changes by ![]() , (or
, (or ![]() ) and the absorption spectrum falls into three groups called branches of the
spectrum. The P branch consists of all transitions with
) and the absorption spectrum falls into three groups called branches of the
spectrum. The P branch consists of all transitions with ![]() :
:
The Q branch consists of all transitions with ![]() :
:
This branch if it is allowed appear at one vibrational transition wavenumber. The R
branch consists of all transitions with ![]() :
:
The intensities of all branches depends both on the population of the vibrational levels and the
magnitude of the corresponding
![]() transitions.
transitions.
The expression for the vibrational-rotational energy term in eq.(58) is given assuming
that the rotational and vibrational movements in the molecule are independent from each other
and can be treated as zero-order approximation. In fact, this approximation is usually not exact
enough in molecular spectroscopy. Particularly, correction of the rotational term by centrifugal
interaction is given in eq. (47). More, quantum mechanical analysis shows that
the rotational constant ![]() and centrifugal distortion constant
and centrifugal distortion constant ![]() in general also depend on
the vibrational quantum number
in general also depend on
the vibrational quantum number ![]() . That is, we have to write
. That is, we have to write ![]() and
and ![]() instead of
instead of ![]() and
and
![]() . In the first approximation assuming that the rotational-vibrational interaction is small
one can write the rotational constant
. In the first approximation assuming that the rotational-vibrational interaction is small
one can write the rotational constant ![]() in the form
in the form
Similarly, the centrifugal distortion constant ![]() can be presented as
can be presented as
The examples for ![]() are given in Table 2 and Table 3
are given in Table 2 and Table 3
In the first approximation the rotation-vibration energy term is usually presented in the form:
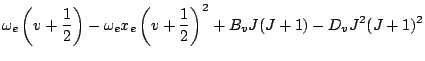 |
(64) |
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.