Eine ganze Reihe von Reaktionen folgt einer Kinetik erster Ordnung; man vermutet deshalb, dass diese Reaktionen über einen geschwindigkeitsbestimmenden unimolekularen Schritt ablaufen. Ein Molekül kann gegebenenfalls duch Stöße mit anderen Molekülen so viel Energie aufnehmen, dass es reagieren kann; Stöße sind aber einfache bimolekulare Ereignisse, die nicht so ohne weiteres zu einer Kinetik erster Ordnung führen.
Die erste erfolgreiche Deutung der unimolekularen Reaktion stammt von Frederik Lindemann und Cyril Hinshelwood. Der Lindemann-Hinshelwood-Mechanismus setzt voraus, dass das Molekül A mit einem Molekül M (z.B. einem Fremdgas-Molekül oder auch einem anderen Molekül A) zusammenstößt und auf Kosten der Energie von M energetisch angeregt wird:
A + M → A* + M, d[A]/dt = k1[A][M].
Das angeregte Molekül kann seine Energie beim Stoß mit einem anderen Molekül verlieren:
A* + M → A + M, d[A*]/dt = - k−1[A*][M].
Das angeregte Molekül kann aber auch von selbst zerfallen und das Produkt B bilden; das heißt, es kann einen unimolekularen Zerfall erleiden:
A* → B, −d[A*]/dt = d[B]/dt = k2[A*]
Wenn der unimolekulare Schritt so langsam ist, dass er geschwindigkeitsbestimmend wird, dann wird er den Gesamtprozess dominieren. So ergibt sich, wie verlangt, für die Gesamtreaktion A → B eine Kinetik erster Ordnung. Man kann das im Detail nachweisen, wenn man die Näherung des stationären Zustandes auf die Bildungsgeschwindigkeiten von A* anwendet:
d[A*]/dt = k1[A][M] − k−1[A*][M] - k2[A*] = 0
Daraus folgt
[A*] = (k1[A][M])/(k2 + k−1[M])
und das Geschwindigkeitsgesetz für die Bildung von B lautet damit
d[B]/dt = k2[A*] = (k1k2[A][M])/(k2 + k-1[M])
Das ist allerdings nicht mehr erster Ordnung. Wenn aber die Geschwindigkeit der Desaktivierung durch A*-M-Stöße sehr viel größer als die Geschwindigkeit des unimolekularen Zerfalls ist, wenn also gilt
k−1[A*][M] >> k2[A*] oder k−1[M] >> k2,
dann können wir im Nenner k2 vernachlässigen und erhalten das Geschwindigkeitsgesetz erster Ordnung:
d[B]/dt = (k1k2[A][M])/(k−1[M]) = {(k1k2)/(k−1)}[M]
Der Lindemann-Hinshelwood-Mechanismus läßt sich leicht überprüfen: Die allgemeine Gleichung für d[B]/dt sagt voraus, dass bei einer Verkleinerung von [M] (das ist in der Praxis eine Erniedrigung des Drucks von M) die Reaktion in eine Reaktion zweiter Ordnung übergehen sollte, denn für k−1[M] << k2 ergibt sich näherungsweise:
d[B]/dt = (k1k2[A][M])/(k2) = k1[A][M].
Physikalisch kommt der Wechsel der Reaktionsordnung dadurch zustande, dass bei kleinen Drücken die bimolekulare Bildung von A* der geschwindigkeitsbestimmende Schritt ist. Schreiben wir das Geschwindigkeitsgesetz in der Form
d[B]/dt = keff[A], keff = (k1k2[M])/(k2 + k−1[M]*),
dann können wir dieses theoretische Ergebnis mit dem Experiment
vergleichen.
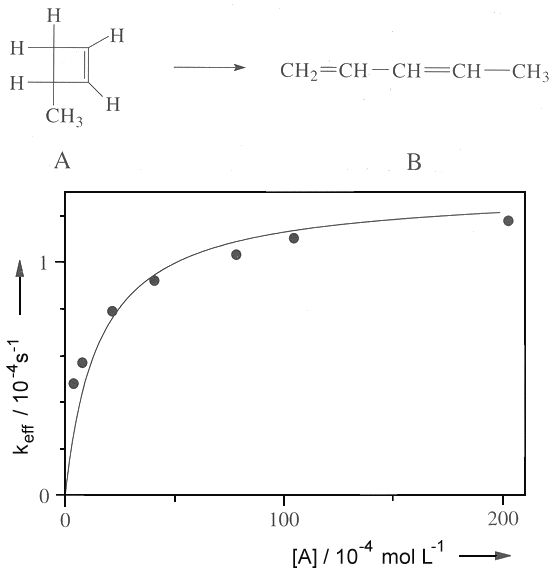 |
| Abb. 1: Experimentelle Daten für die Isomerisierung von 3-Methyl-Cyclobutan (volle Kreise) und deren Vorhersage nach dem Lindemann-Hinshelwood-Mechanismus (Kurve). Werte: k1k2/k-1 = 1,3·10-4s-1, k1 = 0,084 Mol-1s-1. |
Eine Ursache für diese Abweichung liegt u.a. daran, dass der Lindemann-Hinshelwood Mechanismus nicht berücksichtigt, dass bei den meisten Reaktionen eine ganz bestimmte Anregung notwenig ist, damit die Reaktion ablaufen kann. Die Isomerisierung (Abb.1 oben) ist eine Reaktion erster Ordnung; der entscheidende Schritt, das Aufbrechen der (linken) C-C-Bindung ist nur dann möglich, wenn eben diese Bindung in einem hoch angeregten Schwingungszustand vorliegt. Wenn bei einem Stoß eine Anregung erfolgt, so verteilt sich die Anregungsenergie normalerweise über alle Bindungen und über die Rotation des Moleküls, so dass die Isomerisierung erst dann erfogen kann, wenn sich die Energie in der entscheidenden Bindung angesammelt hat. Wir haben also zu unterscheiden zwischen einem angeregten Molekül A*, dessen Anregungsenergie über viele Freiheitsgrade verteilt ist, und einem aktivierten Zustand A#, bei dem die Anregungsenergie so verteilt ist, dass die Reaktion möglich ist. Wir sollten also für den unimolekularen Teil des Mechanismus besser schreiben
A* → A# → P
Nach der Rice-Ramsperger-Kassel-Theorie (RRK-Theorie) hängen
die Zahlenwerte dieser Geschwindigkeitenskoeffizienten mit der Anzahl und
den Frequenzen der zugänglichen Schwingungsfreiheitsgrade zusammen.
Die weiterentwickelte RRKM-Theorie (M steht für Markus) berücksichtigt
auch die Rotation der Moleküle.
Wir wollen uns nun etwas näher mit diesem aktivierten Zustand
A#, dem sogenannten Übergangszustand
beschäftigen
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.