Eine besonders einfache Form nimmt die LCAO-Entwicklung bei homonuklearen zweiatomigen Molekülen an; das sind Moleküle mit gleichen Atomen, wie beispielsweise H2, O2, N2 usw. Es empfiehlt sich, mit dem einfachsten dieser Systeme zu beginnen, dem Wasserstoffmolekül-Ion H2+. Da dieses Molekül-Ion nur ein Elektron enthält, hat seine Betrachtung dieselbe zentrale Bedeutung für Moleküle wie die des Wasserstoffatoms für die Struktur der Atome im Periodensystem.
Dabei beachten wir, dass in den beiden Säkulargleichungen
die Größen αA und αB
gleich sein müssen. Die beiden Enden des Moleküls sind nämlich
nicht unterscheidbar und ein Elektron hat in φA
(an einem Kern) dieselbe Energie wie in φB
(am anderen Kern). Setzten wir folglich αA
= αB = α,
so lauten die Säkulargleichungen:
| cA(a - E) + cB(b
- ES) = 0
cA(b - ES) + cB(a - E) = 0 |
die nur dann eine "chemisch sinnvolle" Lösung (d.h. cA
und cB ungleich Null) besitzen, wenn die Energie die beiden
Werte
| E± = [a ± b]/[1±S] |
annimmt (Herleitung).
Ganz
einfach wird es, wenn S sehr klein gegenüber 1 ist:
| E± @ a ± b |
Bei "etwas größerem S" gilt in besserer Näherung: E± @ a ± (b - aS)
Die Gleichungen zeigen, wie sich ein ursprünglich entartetes Paar
von Energiewerten α in zwei verschiedene Werte
aufspaltet und zwar symmetrisch bezgl. des ursprünglichen Energiewertes,
falls S klein gegen 1 ist. Der eine neue Energiewert liegt dann höher
und der andere niedriger als zuvor. Man erkennt auch, dass die Größe
der Aufspaltung durch den Wert (b-aS)
bestimmt ist. Das MO mit der tiefsten Energie wird bindendes MO
genannt, denn dessen Energie ist kleiner als die des Wasserstoffatoms,
und dieses stellt den Grundzustand des Moleküls dar. Das andere MO
mit der höheren Energie ist das antibindende MO; dessen Energie
ist größer als die des Wasserstoffatoms. Die Ergebnisse sind
im folgenden Bild schematisch dargestellt. Eine Asymmetrie der Aufspaltung
tritt nur dann auf, wenn S nicht gegen 1 vernachlässigt wird.
| E- _____
/ \ _a______/ \___α__ \ / \______/ E+ |
Energieaufspaltung @ ± (b - aS) |
Um nun die Wellenfunktion y
= cAφA + cBφB
für die Energiezustände zu bestimmen, benötigen wir noch
die Koeffizienten cAund cB. Durch Einsetzen der beiden
Energiewerte E± in die Säkulargleichungen erhalten
wir die beiden Lösungen für die Koeffizienten: cA
= ± cB. Die entsprechenden Molekülorbitale sind
dann:
| y+
= cA (φA
+ φB)
y- = cA (φA- fB) |
wobei die cA - wie üblich - über die Normierung (s.u.) bestimmt werden.
| y+
= (φA + φB)/Ö[2(1+S)]
y- = (φA - fB)/Ö[2(1-S)] |
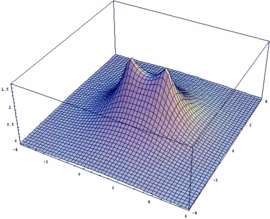
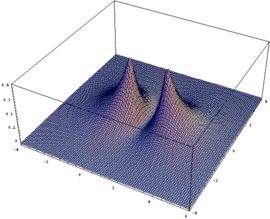
Die sich so ergebenden Wellenfunktionen y+ und y- sowie deren Betragsquadrat |y+|² und |y-|² kann man sich für das H2+-Ion durch anklicken ansehen.
Für den weiteren Verlauf der Rechnung verwenden wir für φA
und φB die Form als 1s-AO für
das Wasserstoffatom; damit können wir dann α,
β
und S für jeden vorgegebenen interatomaren Abstand R berechnen. Auf
diese Weise erhalten wir die elektronische Energie, und wenn wir dazu noch
die Energie der Kernabstoßung e²/4peoR
addieren, so erhalten wir die Energiekurve für das Molekül.
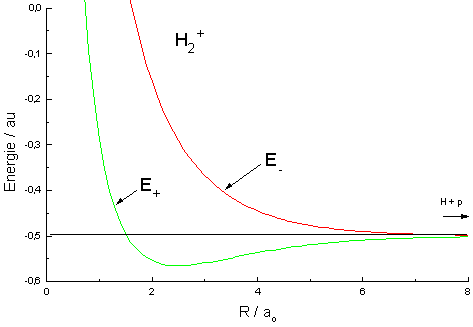 |
| Abb.1: Verlauf der
(absoluten) Energie für das bindende (+) und das antibindende MO (-)
als Fkt. des internuklearen Abstandes R. Alles in atomaren Einheiten: ao = Bohrscher Radius, Energie/au: 1au = zweifache Ionisierungsenergie des Wasserstoffatoms. |
Die Ergebnisse sind in der nebenstehenden Abbildung dargestellt. Die Energiekurve für y+ ist die für den Grundzustand des Moleküls. Aus diesen Energiekurven folgen einige Eigenschaften des Moleküls:
Die Normierung (d.h. unser Elektron im gesamten Raum zu finden) muss 1 sein:
ò |y+|2
= 1 = ∫
cA(φA+
φB)·cA(φA+
φB)
= cA2(
ò
|fA|2 + ∫2|φAφB|
+ ∫ |φB|2
)
¯
¯
¯
1
2S
1
bindend:
cA2 = 1/[2(1+S)]; y+
= (φA + φB)/Ö[2(1+S)]
antibindend: cA2
= 1/[2(1-S)]; y-
= (φA - fB)/Ö[2(1-S)]
Die Wahrscheinlichkeitsdichte P ist danach P = ( |fA|2 +2|φAφB| +|φB|2 )/[2(1+S)], d.h. die "Ladungsdichte des Elektrons" ist pro Kern 1/[2(1+S)] und 1/(1+S) dafür, dass das Elektron zu beiden Kernen gehört und damit die Bindung macht.
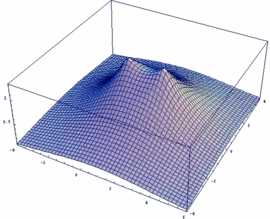
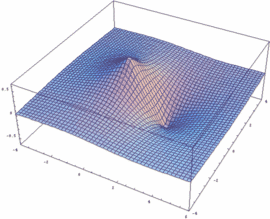
Mathematica-Besitzer können sich leicht selber die Funktionen y+ und y- sowie deren Betragsquadrat |y+|² und |y-|² darstellen (auch für beliebige internukleare Entfernung Rgl):
Rgl=2.4;
psiP=Exp[-Sqrt[x^2+y^2]/2]+Exp[-Sqrt[(x-Rgl)^2+y^2]/2]
psiM=Exp[-Sqrt[x^2+y^2]/2]-Exp[-Sqrt[(x-Rgl)^2+y^2]/2]
Plot3D[psiM, {x,-4,6}, {y,-4,4},
PlotPoints -> 50, PlotRange -> All]
Plot3D[psiP, {x,-4,6}, {y,-4,4},
PlotPoints -> 50, PlotRange -> All]
Plot3D[psiP^2, {x,-4,6},
{y,-4,4}, PlotPoints -> 50, PlotRange -> All]
Plot3D[psiM^2, {x,-4,6},
{y,-4,4}, PlotPoints -> 100, PlotRange -> All]
Der Hamilton-Operator für das H2+ lautet:
H
= - h²/2mD
+e/4peo [-
1/rA
- 1/rB + 1/R]
oder in sogenannten atomaren Einheiten (au): H
= - ½ D - 1/rA
- 1/rB
+ 1/R
Beim H-Problem gilt: ( -½Δ - 1/rA)
φA
= - ½ φA
[Grundzustand Energie in au]
α = òfA*HφAdτ. Aus H-Problem wissen wir: HφA = (- ½Δ - 1/rA - 1/rB + 1/R)φA = - ½ φA + (- 1/rB + 1/R)φA
α = -½ + ∫φA* (- 1/rB + 1/R)φAdτ = -½ + Jmit dem sogenannten Coulombintegral J = - òfA* · 1/rB · φAdτ + 1/R
ß = òfB* HφAdτ = − ½ òfB*φAdτ + ∫ φB* (- 1/rB + 1/R)φAdτ = -½ S + Kmit dem Austauschintegral K = - òfB* 1/rBφAdτ + S/R
und dem Überlappungsintegral
S = - òfB*φAdτ
| E± = [a ± b]/[1±S] = -½ + [J±K]/[1±S] |
Für 1s-Orbitale erhalten wir:
S = e-R (1 + R + R2/3)
J = e-2R (1 + 1/R)
K = S/R - e-R(1
+ R) = e-R[1/R - 2/3R]
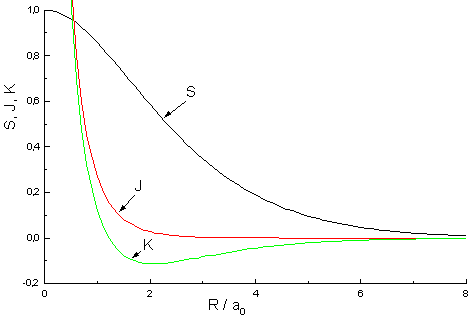 |
| Abb.2: Werteverlauf des Überlappungsintegral S, Coulombintegral J und Austauschintegral K |
Und so leicht findet man das Minimum von E+ mit Mathematica (Ableitung Nullsetzen und nach R auflösen):
DEpl=D[Epl,R];
FindRoot[DEpl==0,{R,{1.5,2.5}}]
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.
{kind=link}
{kind=link}
{kind=link}
{kind=link}