Die Berechnung der Molekülorbitale nach Hückel (Erich Hückel, deutscher Physiker und Theoretischer Chemiker, 1896-1980) soll zunächst in der einfachsten Form dargestellt werden. Für den wahrhaft Interessierten sei auf das Acrolein-Molekül verwiesen.
Wir kennen bereits die LCAO-Näherung, nach
der in der allgemeinen Form gilt:
| Σkck(Hik - E · Sik) = 0 |
Wir machen nun die grobe Näherung Sik = δik, d.h. das Überlappungsintegral zu den Nachbarn wird vernachlässigt, weiterhin setzen wir Hii = α, d.h. es werden nur gleiche Atome (Orbitale) betrachtet und Hik = β für i = k ±1 und Hik = 0 für andere i,k-Werte, d.h. nur die nächsten Nachbarn zum i-ten Atom tragen zur Bindung bei und die Wechselwirkung ist für alle beteiligten Atomorbitale gleich β.
Die Säkulargleichung, die sich aus der obigen allgemeinen Gleichung ergibt lautet dann für ein zweiatomiges Molekül:
(a - E) · c1
+ β · c2
= 0
β · c1 + (a
- E) · c2 = 0
Wir erhalten nur dann nichttriviale Lösungen (d.h. ci¹ 0), wenn die Säkulardeterminante verschwindet:
|
= 0 |
|
= 0 |
Auflösen, d.h. Multiplikation der Hauptdiagonalelemente minus Multiplikation der Nebendiagonalelemente ergibt:
x2-1 = 0 mit der Lösung x1/2 = ± 1,
also E1 = a + b und E2 = a - b.
Im Fall von H2 erhalten wir danach beispielsweise für
den niedrigsten Zustand, der doppelt besetzt ist, Etotal = 2(a
+ b).
Da die Energieabsenkung 2b = -0.174a.u.
beträgt, folgt daraus b = -0.087a.u (=
228 kJ/mol).
Für das (einfach besetzte) H2+-Ion,
Etotal = a + b, erwarten wir danach
eine Energieabsenkung von b = -0.087a.u.(= 228
kJ/mol), was recht gut mit dem wahren Wert von b =
-0.10a.u. (= 262 kJ/mol) überein stimmt.
Für Ethen H2C=CH2 können wir bzgl.
der
π-Bindungen genauso vorgehen. Jedes C-Atom
steuert gerade ein Elektron zum π-System bei,
so dass wir für die totale Energie Etotal = 2a
+ 2b erhalten. Da bei einer Anregung p ®
p* ein Elektron um 2β über den Grundzustand
liegt, können wir für π-Bindungen
b
= -75 kJ/mol abschätzen und die Bindungsenergie kann vorausgesagt
werden.
Für lineares H3+: H-H-H (die H-Atome sind von links nach rechts durchnumeriert) und für ringförmiges H3 lauten die Säkulardeterminanten:
| lineares H3+ |  |
ringförmiges H3+ |  |
||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||
| Ergebnisse | |||||||||||||||||||||||||||
| x1 = 0 | E1 = α | x1/2 = 1 | E1/2 = α - β | ||||||||||||||||||||||||
| x2/3 = ±√2 | E2/3 = α ± β | x3 = -2 | E3 = α + 2β | ||||||||||||||||||||||||
| Etotal = 2(α + β√2) = 2α + 2,83β | Etotal = 2α + 4β | ||||||||||||||||||||||||||
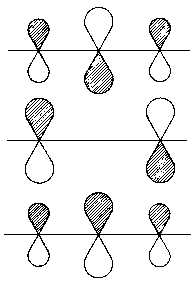 |
| Abb.1: Schematische Darstellung der Orbitale für die Energiewerte a+bÖ2 (energetisch niedrigster Wert, unten), α(Mitte), a-bÖ2 (energetisch höchster Wert, oben). |
| I | Ö2·c1 + 1·c2 + 0·c3 = 0 |
| II | 1·c1 + Ö2·c2 + 1·c3 = 0 |
| III | 0·c1 + 1·c2 + Ö2·c3 = 0 |
Aus I folgt: c2 = -Ö2·c1
Einsetzen in II ergibt: c3 = −c1-Ö2·c2
= −c1 + 2·c1
= c1
Wir erhalten also für das energetisch höchste Niveau:
|
|
|
Den letzen Koeffizienten c1 erhalten wir über die Normierung der Wellenfunktion, wobei wir beachten, dass φi·φk = δik ist:
1 = c1² (1² + Ö2² + 1²) Þ c1 = 1/2
Die Säkulardeterminante von Butadien (C4H6,
CH2=CH−CH=CH2) führt
auf eine Gleichung vierten Grades, wenn wir die C-C-Bindungen betrachten:
|
Die Nullstellen dieser Gleichung sind:
x1 = −½
-
½
Ö5
= −1,618
|
Die Einelektronenergien der vier LCAO-MOs ergeben sich aus der Gleichung
E = a - x β:
| E1 = α + 1,618 β
E2 = α + 0,618 β E3 = α − 0,618 β E4 = α − 1,618 β |
Wir müssen nun unsere 4 Elektronen unterbringen. Die Gesamtenergie des Moleküls ergibt sich als Summe der Energien der besetzten Orbitale. Für das Butadien-Molekül im Grundzustand beträgt die Elektronen-Gesamtenergie daher:
Etotal = 2 (α + 1,618 β) + 2 (α + 0,618 β) = 4α + 4,472 β
Vergleichen wir das Ergebnis mit zwei Ethenmolekülen, für die wir 2· (2α + 2β) = 4α + 4β erhielten, dann sehen wir, dass Butadien um 4,472 b - 4β = 0,472 β ≈ −35 kJ/mol stabiler ist.
Diese zusätzliche Stabilisierung des konjugierten Systems nennt man Delokalisierungsenergie.
Wir können also die energetische Verhältnisse vieler Systeme
relativ leicht abschätzen. Um die Wellenfunktion für die so konstruierten
Zustände zu erhalten, müssen wir die Eigenvektoren bestimmen.
Die zu jedem xi gehörende Eigenvektoren ci
werden wie folgt berechnet: Wir setzten einen Eigenwert in die Säkulargleichungen
ein und erhalten drei unabhängige Gleichungen (eine der vier Gleichungen
ist linear abhängig). Wir lösen diese in Abhängigkeit eines
Koeffizienten, der zuletzt über die Normierung bestimmt wird. (Beispiel
für den ersten Eigenwert x1 = 1,618).
| I | x1c1 + 1·c2 + 0·c3 + 0·c4 = 0 |
| II | 1·c1 + x1c2 + 1·c3 + 0·c4 = 0 |
| III | 0·c1 + 1·c2 + x1c3 + 1·c4 = 0 |
| IV | 0·c1 + 0·c2 + 1·c3 + x1c4 = 0 |
Aus I folgt: c2 = −x1c1 = 1,618 c1
Einsetzen in II ergibt:
Einsetzen in III ergibt:
| 1π: | y1 = c1(φ1 + 1,618 φ2 + 1,618 φ3 + φ4) |
wobei die φi die p-Orbitale von
jedem C-Atom repräsentieren. Analog erhalten wir y2,
y3,
y4,
indem wir x2, x3, x4 statt x1 in die Säkulargleichungen I - IV einsetzen:
| 2π: | y2 = c1(1,618 φ1 + φ2 - f3 − 1,618 φ4) |
| 3π*: | y3 = c1(1,618 φ1 - f2 - f3 + 1,618 φ4) |
| 4π*: | y4 = c1(φ1− 1,618 φ2 + 1,618 φ3 - f4) |
Den letzen Koeffizienten c1 erhalten wir über die Normierung der Wellenfunktion, wobei wir beachten, dass φi·φk = δik ist:
1 = c1² (1,618² + 1² + 1² + 1,618²)
c1 = 0,372
Die Ergebnisse sind in der Tabelle zusammengestellt:
| MO 1 | MO 2
HOMO |
MO 3
LUMO |
MO 4 | |
|---|---|---|---|---|
| Eigenwerte | -1,618 | -0,618 | +0,618 | +1,618 |
| Atom 1 | +0,372 | +0,601 | +0,601 | +0,372 |
| Atom 2 | +0,607 | +0,372 | -0,372 | -0,601 |
| Atom 3 | +0,601 | -0,372 | -0,372 | +0,601 |
| Atom 4 | +0,372 | -0,601 | +0,601 | -0,372 |
| 4π* | 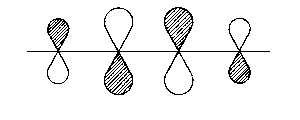 |
überall antibindend |
| 3π* | 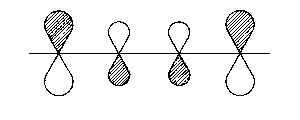 |
1 - 2; 3 - 4 bindend
2 - 3 antibindend |
| 2π | 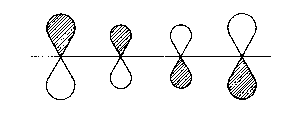 |
1 - 2; 3 - 4 bindend
2 - 3 antibindend |
| 1π | 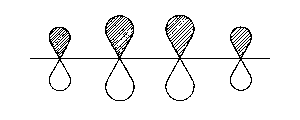 |
überall (1-2-3-4) bindend |
| Abb. 2: Darstellung der Molekülorbitale, wie sich aus Hückels Behandlung des Butadien-Moleküls hervorgehen. Die schraffierten Flächen sollen Bereiche anzeigen, in welchen die Wellenfunktion ein positives Vorzeichen aufweist. | ||
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.