Dank des Doppelspaltexperiments kennen wir bereits ein wesentliches
Prinzip der Quantenmechanik, nämlich dass sich die richtige Wahrscheinlichkeitsamplitude
zur Beschreibung eines Systems als Linearkombination der Einzelamplituden
darstellen läßt. Wir wollen nun molekulare Energiezustände
und Wellenfunktionen bestimmen. Hierzu müssten wir die Schrödingergleichung
Hyn
= En ynlösen,
wobei H der Hamiltonoperator für das
Molekül ist und die ynsind dann
die Molekülorbitale. Zur Analyse des Systems entwickeln wir die Eigenfunktion
yn
nach anderen (uns sehr wohl bekannten) Basisfunktionen φk,
wobei die entsprechenden
φk dann
natürlich nicht mehr Eigenfunktion bzgl. Hyn
sind: y = Σk
ckφk (zur Vereinfachung
ist der Index n weggelassen worden). Der nachfolgende Teil ist mathematisch
abstrakt gehalten. Wer sich jedoch auf etwas einfachere Weise einen ersten
prinzipiellen Eindruck verschaffen möchte, sollte
hier
klicken.
Die auffallendste Eigenschaft eines Molekülorbitals (MO)
ist die Mehrzentrigkeit. Dadurch unterscheidet es sich von einem Atomorbital
(AO). Man beschreibt das Elektron physikalisch am besten, wenn man sagt,
es bewege sich in einem Orbital, das die Umgebung beider (resp.
aller) Kerne umfaßt. Wir wollen zur Beschreibung der Molekülorbitale
y
diese
als Linearkombination von Atomorbitalen
f
darstellen
(LCAO = Linear Combination of Atomic
Orbitals).
Welche f man am besten auswählt, werden
wir bald sehen. Doch zunächst ist die Entwicklung der y-Funktion
nach den φ's mit den üblichen Abkürzungen
für den allgemeinen Fall (beliebiges Molekül) beliebig vieler
φ-Funktionen
in der linken Spalte, bzw. für ein zweiatomiges Molekül (mit
nur 2 φ-Funktionen) in der rechten Spalte der
Tabelle dargestellt:
|
|
|
| y = Σk ckφk | y = cAφA + cBφB |
|
|
|
| Multiplikation mit φi* und ∫ : | Multiplikation mit φA* und ∫ : |
|
|
|
|
Sik = òfi*φkdτ |
SAB = òfA*φB |
|
|
cA(HBA − E · SBA) + cB(HBB − E) = 0 |
Es handelt sich um lineare Gleichungssysteme (Säkulargleichungen), die nur dann eine andere als die mathematisch triviale Lösung ck=0 haben, wenn die sogenannte Säkulardeterminante verschwindet. Im zweiatomigen Fall ist die Lösung auch einfacher zu erhalten. Wir brauchen beide Gleichungen nur nach cA/cB auflösen und gleichsetzen. Vielleicht irritiert es, dass überhaupt eine Größe òfi*φkdτ auftaucht; haben wir doch gelernt, dass die f-Funktionen eines Atoms, also die AOs, orthogonal sind. Stimmt natuerlich, aber eben nur, wenn sie sich auf das gleiche Zentrum (Atom) beziehen. Bei Molekülen sind die AOs unterschiedlicher Atome aber wegen der Mehrzentrigkeit gegeneinander verschoben und da sind sie natürlich nicht zwangsläufig orthogonal. Wir ahnen aber schon, dass es sehr wünschenswert wäre zu wissen, wann das Integral ò fA*φBdτ verschwindet. Wer ungeduldig ist und selber nachdenken möchte, kann schon einmal unterschiedliche Atomorbitale, z.B. 1sA (für Atom A) mit 2px, 2py, oder 2pz des Atoms B "paaren" und sich überlegen, wann das Integral verschwindet. Doch zunächst sei darauf hingewiesen, dass Größen wie HAA, HAB, SAB etc. in allen Anwendungen der LCAO-MO-Theorie auftreten, und deshalb ist es wichtig, ihre physikalische Bedeutung genau zu kennen. Häufig wird auch eine andere Abkürzung gewählt:
αA = HAA = ò fA* HfAdτ αB = HBB = ò fB* HφBdτ
β = HAB= HBA= ò fA* HφBdτ SBA= SAB= S = ∫ fA*φBdτ
Die Gleichungen für das AB-System lauten dann:
cA(αA−
E) + cB(β −
E·S) = 0
cA(β −
E·S) + cB(αB−
E) = 0
Wir wollen uns zunächst die Bedeutung der Größen verdeutlichen: Unter der Annahme einer normierten Funktion φA stellt die Größe αA etwa die Energie dar, die das Elektron haben würde, wenn es sich im Orbital φA am Atom A bewegen würde; eine analoge Interpretation gilt für αB. Die Energie αA ist nicht genau so groß wie EA, die Energie eines Elektrons in φA am isolierten Kern A, denn H enthält auch einen anziehenden Term für den Kern B. αA ist der Erwartungswert der Energie eines Elektrons, das durch φA beschrieben wird. Die Beiträge für αA kommen hauptsächlich aus dem kernnahen Bereich von A, wo auch φA groß ist, und das ist genau der Bereich, in dem H dem Operator eines freien Atoms ähnlich ist. Demnach ist αA (oder αB) etwa die Energie eines Elektrons in φA (oder φB) am Atom A (oder B), die durch die Anwesenheit des zweiten Atoms geringfügig verändert wird. Die Größen αA und αB enthalten Coulomb-Wechselwirkungsterme und werden deshalb kurz Coulombintegrale genannt. Der Betrag der α-Werte steigt, wenn wir für einen bestimmten Orbitaltyp im Periodensystem von links nach rechts gehen. Von links nach rechts nimmt die Elektronegativität oder die "elektronenanziehende Wirkung" der Atome zu.
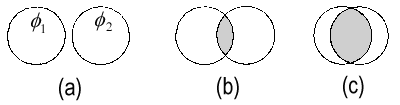 |
| Die Überlappung S12 zwischen zwei gleichen s-Orbitalen φ1 und φ2: (a) S12 ≈ 0; (b) S12 klein; (c) S12 → 1. |
Die obigen Integrale hängen sowohl von φA
als auch von φB ab. Es dürfte
klar sein, dass β und S nur dann dem Betrag
nach zu größeren Werten fähig sind, wenn sich beide Atomorbitale
hinreichend stark überlappen. Wir nennen die Größe S Überlappungsintegral
und die Größe
β Bindungsintegral.
Die Lösung der Säkulargleichungen lautet für homonukleare
Moleküle (bei denen das Atom A mit dem Atom B identisch ist, also
αA
= αB) etwas anders als für heteronukleare
Moleküle (αA ¹aB),
und deshalb werden wir die beiden Fälle gesondert behandeln. Um das
Wesentliche zu verstehen, wenden wir uns zunächst dem einfachsten
Molekül zu: Es ist das H2+-Ion.
Die Laborchemiker mögen nun enttäuscht sein, denn wo begegnet
ihnen schon dieses Ion; doch auch sie können nur vom Einfachen her
das Komplexe verstehen.
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.