First, we want to demonstrate the most simple manner of how Hückel (Erich Hückel, German physicist and theoretical chemist, 1896-1980) calculated molecular orbitals. Anyone truely interested in the method is invited to study Hückel's treatment of the acrolein molecule. We are already familiar with the LCAO approximation. It states, in general:
| Σkck(Hik - E · Sik) = 0 |
We now
In consequence, we employ the following secular equation to describe a diatomic molecule:
(α - E) · c1
+ β · c2 = 0
β · c1 + (α - E) · c2 = 0
We only recieve non trivial (i.e. ci≠0) solutions in cases where the secular determinant becomes zero:
|
|
α-E | β | = 0 | |
| β | α-E |
Division by β and denoting (α-E)/β as x yields
| x | 1 | = 0 | ||
| 1 | x |
The determinant is resolved (multiplication of the elements in both diagonals, subtraction of the product obtained in the secondary diagonal from the product in obtained in the main diagonal)
| x2-1 = 0 <=> x1/2 = ± 1 | |
| or | E1 = α + β and E2 = α - β |
In the case of the molecule H2, we obtain Etotal = 2 (α+β) as state of minimal energy. Two electrons are within this state. The measured reduction of energy in the molecule H2 is E = − 0.174 a.u.. According to the mathematical treatment presented here, this equals 2β, therefore
For the H2+ ion, Etotal = α + β, we expect an energy reduction of
In analogy, the π-bond in ethen H2C=CH2 is to be described. Each carbon atom contributes one electron to the π-system, therefore a total energy of Etotal = 2α + 2β is obtained. In the excited state π → π* of ethen, one electron is 2β above the ground state. Together with spectroscopic measurements, these considerations lead to β = -75 kJ/mol, a value that, in turn, allows to predict the bonding energy.
For the cation H3+, we get the following secular determinants and energy terms:
| linear H3+ |  |
circular H3+ |  |
||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||
| Solutions | |||||||||||||||||||||||||||
| x1 = 0 | E1 = α | x1/2 = 1 | E1/2 = α - β | ||||||||||||||||||||||||
| x2/3 = ±√2 | E2/3 = α ± β | x3 = -2 | E3 = α + 2β | ||||||||||||||||||||||||
| Etotal = 2(α + β√2) = 2α + 2.83β | Etotal = 2α + 4β | ||||||||||||||||||||||||||
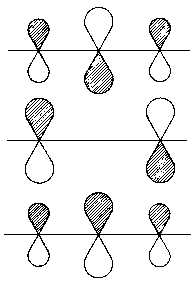 |
| Fig. 1: Representation of the orbitals related the energy value α+b√2 (above, highest energy), α (middle) and α-β√2 (below, lowest energy). |
| I | √2·c1 + 1·c2 + 0·c3 = 0 |
| II | 1·c1 + √2·c2 + 1·c3 = 0 |
| III | 0·c1 + 1·c2 + √2·c3 = 0 |
Equation I establishes the relation c2 = -√2·c1. Inserted in equation II, we get c3 = −c1−√2·c2
= −c1 + 2·c1 = c1. Thus, for the highest energy level, we find the orbital
| π: | Ψ = c1 (Φ1 - √2·Φ2 + Φ3) |
Normalization of the wave function yields the value of the coefficient c1. Note that
1 = c1² (1² + √2² + 1²) ⇒ c1 = 1/2
The secular determinant of butadien (C4H6, CH2=CH−CH=CH2) leads to an equation of the forth degree. The zeros of this equation are:
|
The zeros of this equation are:
x1 = − ½ − ½ √5 = −1.618 x2 = + ½ − ½ √5 = −0.618 x3 = − ½ + ½ √5 = +0.618 x4 = + ½ + ½ √5 = +1.618 |
The single electron energies of the four LCAO molecular orbitals result from equation E = α - x β
|
E1 = α + 1.618 β
E2 = α + 0.618 β E3 = α − 0.618 β E4 = α − 1.618 β |
Four electrons need to be placed. The total energy of the molecule in the ground state is the sum of orbital energies occupied by electrons:
Etotal = 2 (α + 1.618β) + 2 (α + 0.618β) = 4α + 4.472 β
If we compare this result with the 2·(2α + 2β) for two ethen molecules, we come to the conclusion that the energy of butadien is by an amount of [4α-4.472 β] - [4α+4β] = 0.472 β below the energy of two π-bonds. An energy difference of this kind, between a description with located conjugated π-bonds and molecular orbitals obtained in an approach like Hueckel's is called delocalization energy.
Now we are able to estimate the energies of many systems quite easily. To obtain the respective wave functions, we determine the eigenvectors.
To each solution xi of the secular equation, there is an eigenvector ci which is calculated as follows: We insert the eigenvalue xi into the secular equation and recieve three independent equations (one of the four is linear dependent ). We solve this system of equations. The value for the last coefficient emerges when the wave function is normalized. The method is exemplified for the first eigenvalue x1 = 1.618).
| I | x1c1 + 1·c2 + 0·c3 + 0·c4 = 0 |
| II | 1·c1 + x1c2 + 1·c3 + 0·c4 = 0 |
| III | 0·c1 + 1·c2 + x1c3 + 1·c4 = 0 |
| IV | 0·c1 + 0·c2 + 1·c3 + x1c4 = 0 |
Equation I states:
c2 = −x1c1 = 1.618 c1
Inserted in equation II:
c3 = −c1− x1c2 = −c1 + 1.618 · 1.618 c1 = +1.618 c1
Inserted in equation III:
c4 = −c2− x1c3 = −1.618 c1 + 1.618 · 1.618 c1 = c1
The wave function 1π of the respective molecular orbital is a linear combination of the four p-orbitals Φ of the carbon atoms. In an analogous way, the wave functions for orbitals of higher energy Ψ2, Ψ3 and Ψ4 are recieved by inserting the eigenvalues into the secular equations.
| 1π: | Ψ1 = c1(Φ1 + 1.618Φ2 + 1.618Φ3 + Φ4) |
| 2π: | Ψ2 = c1(1.618Φ1 + Φ2 − Φ3 − 1.618Φ4) |
| 3π*: | Ψ3 = c1(1.618Φ1 − Φ2 − Φ3 + 1.618Φ4) |
| 4π*: | Ψ4 = c1(Φ1− 1.618Φ2 + 1.618Φ3 − Φ4) |
Again, normalization yields coefficient c1. Note that Φi·Φk = δik:
1 = c1² (1.618²
+ 1² + 1² + 1.618²)
c1 = 0.372
The following table provides an overview on the recieved coefficients for the butadien molecule for the four eigenvalues. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are denoted.
| MO 1 | MO 2
HOMO |
MO 3
LUMO |
MO 4 | |
|---|---|---|---|---|
| eigenvalue | -1.618 | -0.618 | +0.618 | +1.618 |
| atom 1 | +0.372 | +0.601 | +0.601 | +0.372 |
| atom 2 | +0.607 | +0.372 | -0.372 | -0.601 |
| atom 3 | +0.601 | -0.372 | -0.372 | +0.601 |
| atom 4 | +0.372 | -0.601 | +0.601 | -0.372 |
| 4π* | 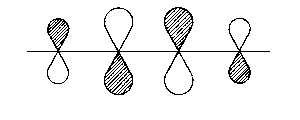 |
throughout antibonding |
| 3π* | 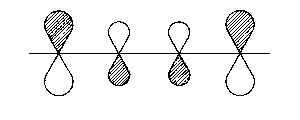 |
1 - 2; 3 - 4 bonding
2 - 3 antibonding |
| 2π | 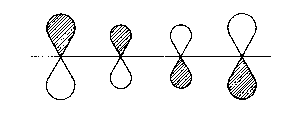 |
1 - 2; 3 - 4 bonding
2 - 3 antibonding |
| 1π | 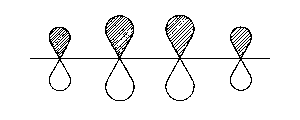 |
throughout (1-2-3-4) bonding |
| Fig. 2: Representation of the molecular orbitals obtained with Hueckel's approach to the butadien molecule. The hatched area shall represent positive values of the wave function | ||
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.