Due to the double slit experiment, we are already familiar with one fundamental principle of quantum mechanics, i.e. that the true probability amplitude for a system can be described as a linear combination of more fundamental single amplitudes. Now we try to determine molecular energy levels and wave functions. Therefore, we need to solve the Schroedinger equation Hψn = En ψn, with the molecule's Hamiltonian operator H and ψn as a representation of the molecular orbitals. For analyzing the system, we expand the eigenfunction ψn by employing well known fundamental functions φk. It is understood that these functions φk are no eigenfunctions with respect to Hψn: y = ∑k ckφk (to simplify, index n has been omitted here). The following explanations treat the problem in an abstract mathematical manner. Anyone who wishes getting a first impression of the principles is invited to click here.
The most conspicious and - when compared with atomic orbitals - distinctive property is that molecular orbitals are polycentric. The best way of describing an electron is to state that it moves within an orbital that is located around both (respectively all) nuclei of the molecule's atoms. Now we want to describe molecular orbitals ψ as a linear combination of atomic orbitals φ (LCAO = Linear Combination of AtomicOrbitals). The appropriate φ is soon to be presented. In a general treatment, first we expand the function ψ for any molecule using functions φ and use the common abbreviations. The equations in the left column refer to the general case where the number of functions φ remains open, whereas the equations in the right column refer to diatomic molecules for which two functions φ are employed.
|
|
|
| ψ = ∑k ckφk | ψ = cAφA + cBφB |
|
|
|
| Multiplication with φi* and ∫: | Multiplication with φA* and ∫: |
|
|
|
|
Sik = ∫φi*φkdτ |
SAB = ∫φA*φB |
|
|
cA(HBA − E·SBA) + cB (HBB−E) = 0 |
We recieved a linear system of equations, the so-called "secular equations". Besides the trivial case with ck = 0, there is only one solution where the secular determinant disappears. For the diatomic molecule, the problem is easily solved by transforming both equations to have cA/cB on the left side and equate both equations of the system. Maybe it is confusing to obtain the term ∫φi*φkdτ as we have learned that the functions, i.e. the atomic orbitals, are orthogonal. This still holds true, but in contrast to AOs of single atoms, in our case, each AO has a centre of its own because there is a shift in position. In consequence, they are not necessarily orthogonal.
We already anticipate that it would be desirable to know the case where the integral ∫ φA*φBdτ disappears. Anyone who wants to consider the problem for himself in advance may combine an atomic orbital of atom A, e.g. 1sA with atomic orbitals like 2px, 2py or 2pz of atom B and pay attention to the case where the integral becomes zero. But be aware that HAA, HAB, SAB etc. appear in all applications of the LCAO-MO-theory and thus it is indispensible to know their physical meaning. Often, you will find another representation:
αA = HAA = ∫φA* HφAdτ
αB = HBB = ∫φB*
HφBdτ
β = HAB = HBA = ∫φA*
HφBdτ
SBA = SAB = S = ∫φA*φBdτ
Using this notation, the equations for the AB-system become:
cA(αA−E) + cB(β−E·S) = 0
cA(β−E·S) + cB(αB−E) = 0
First, let's focus on the meaning of involved quantities: Under the assumption of a normalized function φA the αA represents roughly the energy of an electron imagined to move in orbital φA of atom A; an analogous interpretation applies to αB. Energy αA is below EA for the wave function φA of an electron bound solely to nucleus A because in H, there is also a component that represents attraction to nucleus B. αA is the expected value for the energy that is described by φA. Especially the sphere close to nucleus A contributes to αA where φA is large too, and this is as well the sphere where H resembles the operator of the unbound atom. Thus αA (oder αB) is approximately the energy of an electron in φA (or φB) close to nucleus A (or B) which is slightly changed by the presence of the other atom. As αA and αB contain terms for Coulomb's forces, they are called the coulombic integral. For a certain type of orbital, the absolute value of α increases from the left to the right for the elements of one row in the periodic table of elements. Note that the same increase is found for the atom's electronegativity or "attraction" towards electrons.
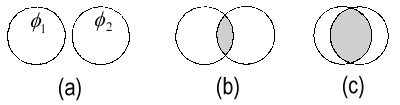 |
| The overlap S12 between two identical s-orbitals φ1 and φ2: (a) S12 ≈ 0; (b) small S12; (c) S12 →1. |
The integrals above depend both on φA and φB. Obviously, the values for β and S are strongly dependent on possible overlap between the two atomic orbitals. S is called the overlap integral and β resonance integral . The solution of the secular equations for homonuclear molecules (consisting identical atoms A and B) with αA = αB differs from that for heteronuclear molecules (αA ≠αB). Thus, we treat the two cases independently. To comprehend the essence, we first focus on the most simple molecule - the H2+-Ion. Maybe, the laboratory chemist is disappointed now as he never faces this ion. But even he won't be able to treat complicated molecules without an understanding for the basic ones.
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.