

Next: Abschließende Bemerkungen (für Fortgeschrittene)
Up: Die Born-Oppenheimer-Näherung
Previous: Entkopplung der Elektronenbewegung
Die Funktion
W'(R) in Gln. (18) nennt man
``Potentialhyperfläche,'' oder bei zweiatomigen Molekülen ``Potentialkurve.''
Die ``adiabatische Korrektur''
A(R) ist im Grunde genommen relativ
leicht zu berechnen [3], man
verzichtet in der Praxis jedoch meistens darauf
(d.h. man setzt
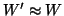 )
da
A(R) so klein ist,
daß die übrigen Fehler meist viel größer sind.
Um eine Vorstellung von der Größe von
A(R) zu bekommen, kann man sich
überlegen, daß die zweite Ableitung der elektronischen Wellenfunktion
)
da
A(R) so klein ist,
daß die übrigen Fehler meist viel größer sind.
Um eine Vorstellung von der Größe von
A(R) zu bekommen, kann man sich
überlegen, daß die zweite Ableitung der elektronischen Wellenfunktion
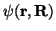 nach einer Kernkoordinate im wesentlichen gleich der zweiten
Ableitung nach einer Elektronenkoordinate ist, welche schließlich mit dem
Kehrwert der Kernmasse gewichtet wird.
nach einer Kernkoordinate im wesentlichen gleich der zweiten
Ableitung nach einer Elektronenkoordinate ist, welche schließlich mit dem
Kehrwert der Kernmasse gewichtet wird.
Abbildung 1:
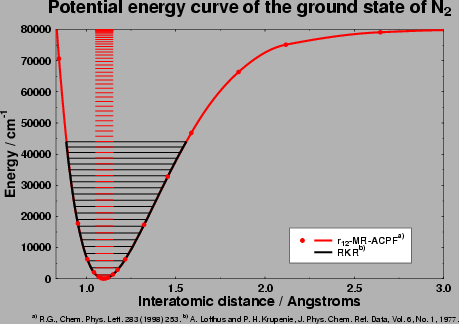 |
Die Abbildung 1 zeigt als Beispiel die Potentialkurve des
(elektronischen)
Grundzustands des Stickstoff-Dimeren (N2) [4].
Man sieht, daß in der Nähe des Gleichgewichtsabstandes
(
Re=1.097685Å [5])
W(R) ungefähr quadratisch ist.
Zu kleineren Abständen steigt die Potentialkurve schnell an, während sie
zu größeren Abständen hin zum Dissoziationslimit (
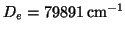 [5]) strebt.
[5]) strebt.
Abbildung:
Rotierendes zweiatomiges Molekül
(schematisch)
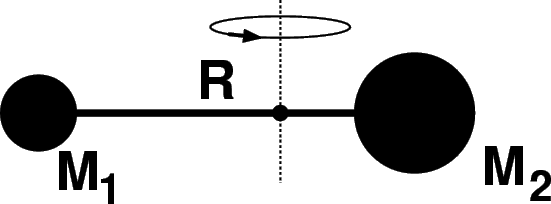 |
Im Fall eines zweiatomigen Moleküls (siehe Abbildung 2)
schreibt man üblicherweise die
Kern-Schrödinger-Gleichung (18) in der Form
![\begin{displaymath}
\left[
{1\over 2\mu}
\left(
-{d^2\over d R^2}
\;+\; {J(J+1)\over R^2}
\right)
\;+\; W'(R)
\;-\; E
\right] \xi(R) \;=\; 0
\end{displaymath}](img42.gif) |
(19) |
Hier ist R der Abstand der beiden Atomkerne zueinander,
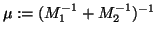 ist die reduzierte Masse und
ist die reduzierte Masse und
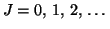 ist die Rotationsquantenzahl.
Im Fall des Grundzustands von N2 gibt es ca. 60 Vibrationszustände.
ist die Rotationsquantenzahl.
Im Fall des Grundzustands von N2 gibt es ca. 60 Vibrationszustände.
Die ro-vibronischen Energieterme eines zweiatomigen Moleküls lassen sich
schreiben als
| E(v,J) |
= |
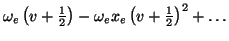 |
|
| |
|
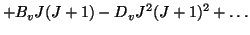 |
(20) |
mit den Rotationskonstanten
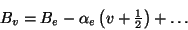 |
(21) |
und den Anharmonizitätskonstanten
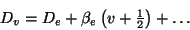 |
(22) |
Die Gln. (20) läßt sich auch formal in Form der Dunham-Reihe
schreiben
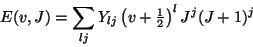 |
(23) |
wobei die Ylj mit den entsprechenden Molekülkonstanten zu identifizieren
sind.
Next: Abschließende Bemerkungen (für Fortgeschrittene)
Up: Die Born-Oppenheimer-Näherung
Previous: Entkopplung der Elektronenbewegung
Robert Gdanitz
1999-07-05
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.