Zeeman Effect
We will consider the influence of a positively charged nucleus on the movement of an electron. In the classical model,the electron (Bohr atom model) is moving along a
closed path. The current arising from this motion is as follows I =
−e/τ (τ: orbital period). The
magnetic moment ![]() e/τ (π r²). And for the kinetic moment L =
mevr = me2pr²/τ where v=2πr/τ
e/τ (π r²). And for the kinetic moment L =
mevr = me2pr²/τ where v=2πr/τ
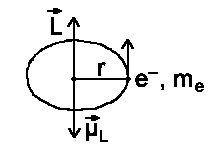 |
Next we obtain:
Since the kinetic moment is represented by an operator in quantum mechanics, we
obtain the eigenvalues µLz of z-component (Lz y =
ml h y):
µLz = − e/2meh·
ml where
µB = eh/2me (Bohr
Magneton)
| µLz = −µB· ml
where µB =
e |
µB =
eh/2me = 9,273 · 10-24 J/K = 5,656 .
10-5 eV/T
= 1,4 · 1010 Hz/T (T:
Tesla)
The energy of such a system in the magnetic field ![]() is as follows:
is as follows:
| EB = −µLz· B = µB · ml· B |
and the total energy is E = E0 + EB , i.e. the energy
levels having a kinetic moment with quantum number l are split into
2l + 1 new levels:
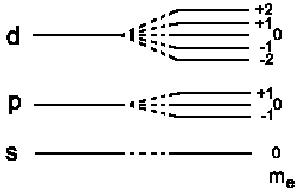 |
This effect is also referred to as ordinary Zeeman Effect |
The hydrogen ground state (ml = 0) stays uninfluenced. This is
not accidental because H is a paramagnetic element! The reason so is because of
electron spin. Now we consider the spin in classical mechanics as rotating
around the axis electron. We also find here
| µSz = −gS µB ·
ms EB = gS· µB· ms· B |
The so-called gyromagnetic factor gS is obtained from the relativist Dirac-equation with gS = 2. Experiments give:
gS = 2,00231930438(6)
This value can be explained by quantum electrodynamics which is
the full quantum theory of electromagnetic field. Moreover, the
electromagnetic field is expanded into normal vibrations which are quantized like a harmonic oscillator. Additionally, each normal vibration has its zero energy
point hw/2. This means there are fluctuating electromagnetic
fields even if there is no influence from external fields. Though the
average field is zero, the squared average field isn't equal to 0. This
leads to the squared average fluctuations of positions.
One can find more detailed information about the Zeeman effect here.
The value gS · µB≈ 28 GHz/T shows us which energy state is higher according to
electron spin interaction with magnetic field. Since now it's possible to
produce magnetic fields with the strength of a few Teslas, we expect to detect
transitions in GHz region (microwaves) when applying such fields. That's why
Electron-Spin-Resonance (ESR)
Spectroscopy is involved with microwaves. When applying NMR
method we have a deal with MHz region. We will discuss it in more detail later.
Spin-Orbital Coupling
Two possible orientations of electron spin (![]() ) relative to orbital
moment (
) relative to orbital
moment (![]() )
give rise to energy level doubling (this is not true for s-levels) which
are spectroscopically resolved since the spectral lines are detected as a pair
of lines (doublet) when one has enough resolution. The most well-known example is
the Na D-line at 5890 Å and 5896 Å wavelengths. To understand the physics of
spin-orbital coupling (or interaction) lets take a look at the atomic nucleus:
the electron revolves around the nucleus and has an electronic orbital moment
)
give rise to energy level doubling (this is not true for s-levels) which
are spectroscopically resolved since the spectral lines are detected as a pair
of lines (doublet) when one has enough resolution. The most well-known example is
the Na D-line at 5890 Å and 5896 Å wavelengths. To understand the physics of
spin-orbital coupling (or interaction) lets take a look at the atomic nucleus:
the electron revolves around the nucleus and has an electronic orbital moment ![]() .
The electron produces a magnetic field
.
The electron produces a magnetic field ![]() that is parallel to
that is parallel to ![]() (because of the
positive nucleus charge). Since the electron in disucussion is
at rest, we have only the interaction between the magnetic
field and the magnetic moment
(because of the
positive nucleus charge). Since the electron in disucussion is
at rest, we have only the interaction between the magnetic
field and the magnetic moment ![]() S
of spin. This interaction is proportional to
S
of spin. This interaction is proportional to ![]() S·
S· ![]() .
Since
.
Since ![]() ||
||![]() and
and ![]() S ||
S ||![]() the interaction is
proportional to
the interaction is
proportional to ![]() ·
· ![]() , that explains the spin-orbital coupling. According to this type of interation, the electron
energy is as follows.
, that explains the spin-orbital coupling. According to this type of interation, the electron
energy is as follows.
ESL = cSL· ![]() .
. ![]() /
/h²
where cSL is the proportionality constant which can be derived from the Dirac equation.
And what about ![]() .
. ![]() ? Now we have
the total kinetic moment
? Now we have
the total kinetic moment ![]() as observable value:
as observable value:
![]() =
= ![]() +
+ ![]()
We can't set a vector randomly in quantum mechanics. BY squaring the above equation, we get:
J² = L² + S² + 2 ![]() .
. ![]() ®
® ![]() .
. ![]() = ½ (J² − L² − S²)
= ½ (J² − L² − S²)
Since the squared kinetic moment is a conservative quantity we can substitute the quantum mechanical operators with eigenvalues (we will now use the small letter for eigenvalues):
![]() .
. ![]() º L. S = ½ [J(J + 1) −
L(L + 1) − S(S + 1)]
º L. S = ½ [J(J + 1) −
L(L + 1) − S(S + 1)] h²
We now obtain for the energy of spin-orbital coupling:
ESL = cSL/2[J(J + 1) − L(L + 1) − S(S + 1)]
The value cSL can be directly observed in the doublet structure of the optical spectra.
For H-atom one can obtain (J = L ± S, S = ½)
ESL ( ≡ J = L + ½) = ½ cSL · L
ESL (¯ ≡ J = L − ½) = − ½ cSL (L + 1)
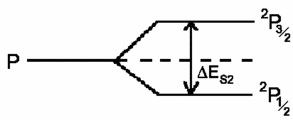 |
J = L + S ≡ 1 + ½ = 3/2 |
| J = L − S ≡ 1 − ½ = ½ |
The separation between two levels is as follows
ΔESL = ESL() − ESL (¯) = ½ cSL (2L + 1)
 |
Together with proportionality constant cSL Dirac obtained the energy separation corresponding to spin-orbital interaction:
ΔESL = |En| Z²α²/n l(l +1) ≈ 5,3 · 10-5 Z²/n l(l + 1) |En|
where α = e²/4πh²ε0c =
1/137,0359895 is the Sommerfeld fine
structure constant.
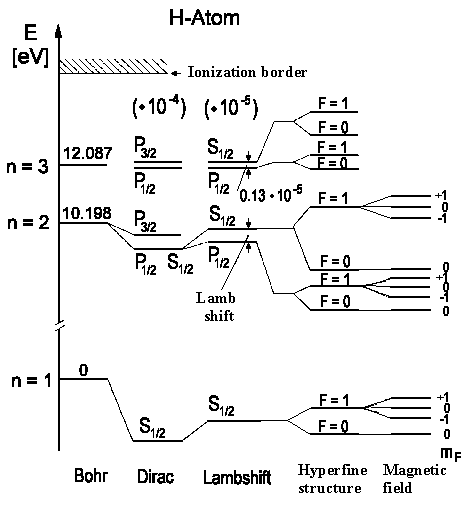
The energy eigenvalues (spin-orbital + relativistic effects) for H-atom are obtained from the full Dirac theory:
EnJ = − mc²/2α²Z²/n²{1 + α²Z²/n(1/J + ½− ¾ n)}
According to this theory, energy levels having the same J and n values are degenerate. For instance, states 2²S½ and 2²P½ would have the same energy. Lamb and Rutherfordproved in 1947 that this degeneracy doesn't correspond to what is observed experimentally. They excited the transition ²S½ ← ²P½ using radiospectroscopy i.e. the energy levels are split and are not degenerate. Moreover this splitting is only about 0,03528 cm-1.
However it's not all concerning level
splitting: there is one more spectral line splitting, the so-called
hyperfine structure. It's caused by interaction between magnetic
field (from electron movement) and nuclear spin. For instance, the hydrogen atom
has one proton with spin I = ½ and corresponding magnetic moment ![]() P = −gI
e
P = −gI
eh/2mp![]() /
/h = µK![]() /
/h
Since a proton is heavier than an electron, its magneton µK
(µK = gI eh/2mp) is lighter by a factor
me/mp !
Similar to fine structure coupling, we must connect ![]() and
and ![]() now having "new" total
kinetic moment:
now having "new" total
kinetic moment:
![]() =
= ![]() +
+ ![]()
where Fy =
F(F +1) h² y and Fzy
= mF hy ; mF = −F,
−F+1, ... F−1, F
Squaring the equation for ![]() and solving it
relatively
and solving it
relatively ![]() .
. ![]() one can obtain the
hyperfine energy structure:
one can obtain the
hyperfine energy structure:
EHFS = cHFS/2{F(F + 1) - J(J + 1) - I(I + 1)}
The lowest term of hydrogen 1²S½ is then split into two terms with F = 1 (spin electron and spin proton ) and F = 0 (spin electron and spin proton ¯ antiparallel). The transition between these two levels can only occur when the spin turns over. This, however, is a very rare phenomenon (it, on average, happens once every 10 million years in a hydrogen atom) which is not observable in our Universe. It lies near n» 1,4204 GHz (λ = 21 cm).
Since this transition changes the system kinetic moment from F =
1h into F = 0h but from the fact
that the total kinetic moment should be the same follows photon must take the
hydrogen atom kinetic moment. And so a photon also possesses kinetic moment.
However, the photon spin is as follows
| SPhoton =
1 |
The proton spin has interesting consequences for optical spectra since we
obtain the selection rule for transitions. We will have for the
final total kinetic moment ![]() f of the system
that was originally at some initial state
f of the system
that was originally at some initial state ![]() i
according to the kinetic moment conservation law:
i
according to the kinetic moment conservation law:
![]() i =
i = ![]() f +
f + ![]() Photon
Photon
Certainly ![]() Photon shouldn't be necessarily equal to
Photon shouldn't be necessarily equal to ![]() because photon can also
possess (together with spin) the orbital moment relative to an atom. The photon
emission with unchanged orbital moment visually means that photon has left the
boundary areas. Nevertheless, this process is very improbable so that quantum
numbers Ji and Jf have the following relation according to
the kinetic moment conservation law:
because photon can also
possess (together with spin) the orbital moment relative to an atom. The photon
emission with unchanged orbital moment visually means that photon has left the
boundary areas. Nevertheless, this process is very improbable so that quantum
numbers Ji and Jf have the following relation according to
the kinetic moment conservation law:
| ΔJ = Jf − Ji = 0 , ±1 |
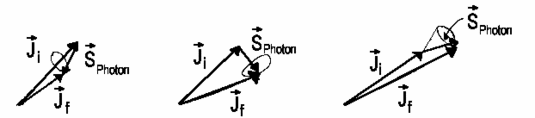 | ||
| ΔJ = −1 ΔM = 0 , ±1 P-Branch |
ΔJ = 0 ΔM = ±1 Q-Branch |
ΔJ = +1 ΔM = 0 , ±1 R-Branch |
If Jf = 0 and Ji = 0 then ΔJ=0 is
forbidden according to the kinetic moment conservation law. If there is small
interaction between ![]() and
and ![]() then there is special selection rule for spin that states:
then there is special selection rule for spin that states:
| ΔS = 0 |
 |
The selection rule for electron orbital moment has been given here without
any derivation:
| Δl = ±1 |
The possible transitions are shown on the illustration to the left (Balmer
a-Line, n = 3 ↔ n = 2). Since H atoms move there
are Doppler distribution of these optical lines, that can be resolved by using
Doppler free spectroscopy which can help us to prove the theory.
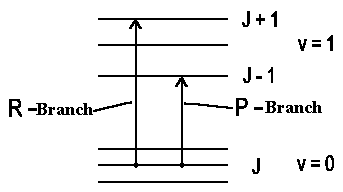
Certainly we must also have kinetic moment conservation for
increasing rotation of a molecule. In the simplest case of a linear molecule for
which electron contribution into kinetic moment is
small so that ΔJ = ±1, i.e. the rotation can be increased by
1h or decreased by 1h when
photon is absorbed. It's said about P-Branch (the rotation is decreased by
1h) and R-Branch (the rotation is increased by
1h).
If total electron kinetic moment isn't equal to 0 or if we have a deal with a non-linear molecule then the kinetic impulse vector character according to the selection rule undergoes some changes, i.e. there is also a transition DJ = 0 (Q-Branch) possible (when J = 0 → J = 0 !).
![]()
Auf diesem Webangebot gilt die Datenschutzerklärung der TU Braunschweig mit Ausnahme der Abschnitte VI, VII und VIII.